Wie werden Arzneimittel im Korper verteilt? Wie gelangen sie an den Wirkort und docken an die richtigen Bindungsstellen an? Wie werden sie im Korper metabolisiert und schlie?lich wieder ausgeschieden? Welches sind die Mechanismen, die diesen Transportprozessen zu Grunde liegen? All dies sind essentielle Fragen der Pharmakokinetik, eines der wichtigsten Gebiete der Arzneimittelforschung und -entwicklung.
In didaktisch geschickter Form beantwortet das vorliegende Lehrbuch diese Fragen, und besticht zusatzlich durch sein ansprechendes Layout mit durchgehend vierfarbigen Abbildungen.
Nicht nur ein Muss fur alle fortgeschrittenen Studenten der Pharmazie, Medizinalchemie, Chemie, Pharmakologie und angrenzender Disziplinen, sondern auch ein wertvoller Begleiter fur Dozenten und Mitarbeiter der pharmazeutischen Industrie.
Aus dem Inhalt:
* Der Weg des Wirkstoffs zum molekularen Target
* Gastrointestinale Absorption
* Prozesse an der Leber
* Eliminierung an der Niere
* Verteilung ins Gehirn: die Blut-Hirn-Schranke
* Verteilung zur Plazenta
- German
- ePUB (handyfreundlich)
- Über iOS und Android verfügbar
eBook - ePub
Über dieses Buch
375,005 Studierende vertrauen auf uns
Zugang zu über 1,5 Millionen Titeln zu einem fairen monatlichen Preis.
Mit unseren Lerntools kannst du noch effizienter lernen.
Information
1
Einleitung
1.1 Übersicht über das Thema Pharmakokinetik
In der modernen Arzneimitteltherapie und -forschung gilt der Grundsatz, dass ein pharmazeutischer Wirkstoff an eine molekulare Zielstruktur binden muss, damit ein biologischer Effekt ausgelöst werden kann. Unter Zielstrukturen, auch Targets genannt, werden Rezeptoren, Enzyme, Ionenkanäle und Transporter zusammengefasst. Die Rezeptortheorie (Maehle et al., 2002) wurde durch Paul Ehrlich mitbegründet, der zu Beginn des 19. Jahrhunderts postulierte, dass bestimmte chemische Strukturen auf der Zelle für die selektive Bindung von Substanzen verantwortlich sind und dass die „Medikamente nicht wirken, wenn sie nicht gebunden sind“ (Corpora non agunt nisi fixata, Ehrlich, 1909, S. 214):
„Beherrscht wird das ganze Gebiet von einem ganz einfachen, ich möchte sagen selbstverständlichen Grundsatz. Wenn in der Chemie das Gesetz gilt: corpora non agunt nisi liquida, so ist für die Chemotherapie maßgebend: corpora non agunt nisi fixata!“
Für Enzyme beschrieb Emil Fischer 1894 das Schlüssel-Schloss-Prinzip am Beispiel der spezifischen Bindung zwischen Enzym und Substrat (Fischer, 1894):
„Um ein Bild zu gebrauchen, will ich sagen, dass Enzym und Glucosid wie Schloss und Schlüssel zu einander passen müssen, um eine chemische Wirkung auf einander ausüben zu können.“
Eine nicht-kovalente Wechselwirkung führt zu einem Komplex zwischen Substrat und Enzym, dessen relative Bindungsstärke man als Affinität bezeichnet. Um der konformationellen Flexibilität der beiden Bindungspartner Rechnung zu tragen, wurde schließlich das Induced-Fit-Konzept („Hand-im-Handschuh-Prinzip“) geprägt (Koshland, 1958).
Ein Beispiel für ein Enzym als Zielstruktur ist die Neuraminidase des Grippevirus, welches sich in den Epithelzellen der Atemwege repliziert. Der antivirale Wirkstoff Oseltamivir (Tamiflu®), der bei der medikamentösen Therapie der Grippe verwendet wird, bindet an die virale Neuraminidase und hemmt dadurch ihre Funktion. Dies führt dazu, dass sich das Virus nach der Replikation nicht mehr von der Wirtszelle lösen und sich deshalb nicht weiter vermehren kann (Wutzler & Vogel, 2000). Die Bindung von Oseltamivir an die Neuraminidase ist in Abbildung 1.1 dargestellt.
Abb. 1.1 Damit ein pharmazeutischer Wirkstoff einen biologischen Effekt auslösen kann, muss er an ein molekulares Target binden. Die Abbildung zeigt den antiviralen Wirkstoff Oseltamivir (Tamiflu®, gelb), gebunden an das Enzym Neuraminidase N1 des Influenza-Virus A vom Subtyp H5N1. Rot eingezeichnet sind die mutmaßlichen inter molekularen Wechselwirkungen, welche der Wirkstoff mit dem Enzym eingeht. Die Bindung blockiert die Active Site des Enzyms und damit dessen Funktion und verhindert die Vermehrung des Virus (Koordinanten aus Russell et al., 2006; Bild erstellt mit PyMOL, http://pymol.sourceforge.net).
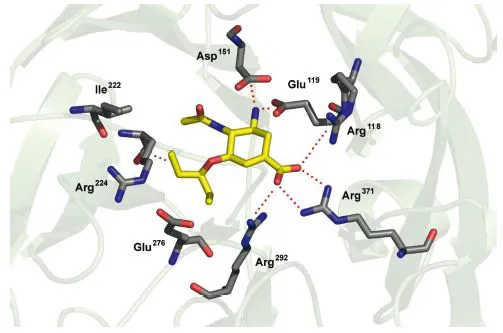
Neben der Bindungsstärke, der Affinität zum Target, ist die Selektivität ein weiterer wichtiger Aspekt. Der Wirkstoff soll nur an ein definiertes Target binden (z.B. an Proteine des Grippevirus), nicht aber an andere Rezeptoren (z.B. körpereigene Proteine). Eine unselektive, d.h. gleichzeitige Hemmung verschiedener Targets kann zu Toxizität und unerwünschten Wirkungen führen.
Eine starke und selektive Bindung eines Wirkstoffs an eine Zielstruktur ist aber nur eine Voraussetzung für einen biologischen Effekt und damit eine effektive medikamentöse Therapie. Eine weitere essentielle Bedingung ist, „dass der Wirkstoff den Wirkort überhaupt in ausreichender Konzentration erreicht“ (Clark, 1933). Oseltamivir beispielsweise wird peroral in Form von Kapseln verabreicht und muss aus dem Darm zu den Epithelzellen der Atemwege und zu den Grippeviren gelangen.
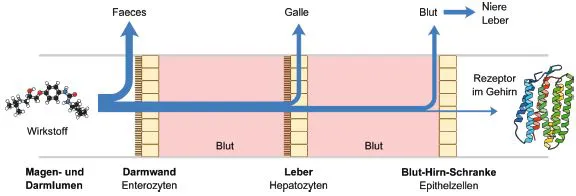
Einem Wirkstoff – oder der Gesamtheit aller Wirkstoffmoleküle aus der Arzneiform – stellen sich auf dem Weg aus dem Darm zur Zielstruktur (z.B. der Lunge) zahlreiche Barrieren in den Weg, welche den Transport behindern und die Menge des Wirkstoffs, der schließlich das Target erreicht, signifikant reduzieren können (s. Abb. 1.2, van de Waterbeemd et al., 2001). Unter Barrieren versteht man Transportbarrieren (z.B. Membranen, Efflux-Proteine), physikochemische Barrieren (z.B. Löslichkeit, pH) und biochemische Barrieren (z.B. metabolisierende Enzyme, Plasmaprotein-Bindung). Bereits der Zerfall und die Freisetzung (Liberation) des Wirkstoffs aus der Arzneiform im Darm stellen eine erste Barriere dar. Der Wirkstoff muss im Darm in Lösung gehen, damit er absorbiert werden kann. Hierbei spielen u.a. die Arzneiform (Voigt, 2000), der Inhalt des Magen- Darm-Trakts und die physikochemischen Eigenschaften des Wirkstoffs eine entscheidende Rolle (Hörter & Dressmann, 2001). Wird der Wirkstoff unvollständig aus der Arzneiform freigesetzt, kann nur ein Teil der gesamten Dosis den systemischen Kreislauf und damit die molekulare Zielstruktur erreichen. Säurelabile Wirkstoffe können zudem im sauren Milieu des Magens (pH 1 bis 2) abgebaut werden (z.B. Penicilline, Digoxin, Erythromycin, Protonenpumpen-Inhibitoren). Im Darm kann der Wirkstoff bereits bevor er das Target erreicht, biotransformiert werden und seine Aktivität verlieren (Doherty & Charman, 2002).
Abb. 1.2 Die Abbildung illustriert Beispiele von Barrieren, die sich einem Wirkstoff auf dem Weg zur molekularen Zielstruktur in den Weg stellen. Nur ein kleiner Teil der verabreichten Dosis erreicht tatsächlich das molekulare Target, welches sich hier im Gehirn befindet.
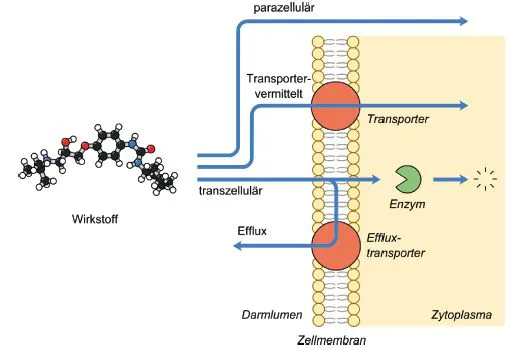
Eine wichtige Barriere ist die Transportbarriere. Wirkstoffe müssen nach der oralen Verabreichung auf dem Weg zu ihrem Target in der Regel mehrere Zellschichten überwinden. Die erste Zellschicht, auf die der oral verabreichte Wirkstoff trifft, ist das einschichtige Oberflächenepithel im Darm, das von den Darmzellen (Enterozyten) ausgebildet wird (Welsch, 2006). Der Prozess der Aufnahme des Wirkstoffs aus dem Darm in die Blutgefäße der Darmzotten wird alsintestinale Absorption bezeichnet. Die verschiedenen möglichen Transportwege an der apikalen Enterozytenmembran sind in Abbildung. 1.3 dargestellt. Wirkstoffe können zwischen den Zellen passieren (sog. parazellulärer Transport). Dieser Transportweg spielt aber eine untergeordnete Rolle und ist auf hydrophile Wirkstoffe mit einem Molekulargewicht von < 200 g/mol beschränkt (z.B. Aciclovir, Paracetamol), da die Zellen in den Zwischenräumen durch Verschlusskontakte (Tight Junctions) eine Barriere gegen den freien Stofftransport ausbilden (z.B. Schnee-berger & Lynch, 2004). Die meisten Wirkstoffe passieren die Zelle transzellulärund müssen dabei hydrophobe Zellmembranen überwinden. Hier sind verschiedene Transportmechanismen möglich. Ein lipophiler Wirkstoff kann passiv über die Membran diffundieren. Für hydrophile und geladene Wirkstoffe ist die Zellmembran – die im inneren Kern hydrophob ist – jedoch eine Transportbarriere. Die physikochemischen Eigenschaften eines Wirkstoffs determinieren deshalb sein Verhalten gegenüber der Transportbarriere.
Abb. 1.3 Die Abbildung zeigt, über welche Transportwege ein pharmazeutischer Wirkstoff die apikale Zellmembran eines Enterozyten im Darm passieren kann.
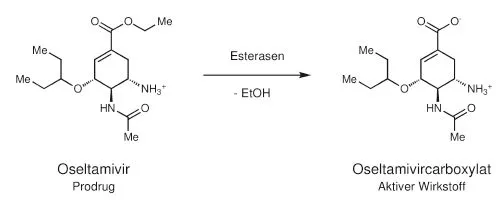
Oseltamivir beispielsweise ist nur durch einen chemischen Kunstgriff oral absorbierbar. Der Wirkstoff wird nämlich erst nach der Absorption in den Enterozyten und in der Leber durch enzymatische Hydrolyse freigesetzt (s. Abb. 1.4). Die Umwandlung erfolgt nahezu vollständig. Es handelt sich um einen sogenannten Prodrug, ein Begriff, der 1958 von Adrien Albert geprägt wurde:
„Sometimes the substance as administered, is only a ,pro-drug’ which has to be broken down to give the true drug.“ (Albert, 1958, S. 421).
Die polare und im Magen-Darm-Trakt positiv geladene Carboxylatgruppe des aktiven Wirkstoffs wurde mit Ethanol verestert, damit der Wirkstoff intestinal absorbiert wird und als Kapsel oral verabreicht werden kann (Li et al., 1998).
Der Körper ist auf effiziente Mechanismen angewiesen, die auch hydrophile und geladene Nahrungsbestandteile die Membranbarriere passieren lassen, z.B. Ionen, Nukleinsäuren, Aminosäuren, Peptide, Lipide oder Zucker. Das wird mit Transportproteinen, d.h. Transportern und Kanälen, erreicht, die den selektiven Transport bestimmter Substanzen und Substanzgruppen über Zellmembranen durch verschiedenartige Mechanismen begünstigen. Glucose beispielsweise kann aufgrund ihrer hydrophilen Hydroxylgruppen Zellmembranen nicht passiv überwinden. Der Zucker wird deshalb über die Transporter GLUT (Glucose- Transporter, z.B. an der Blut-Hirn-Schranke) und SGLT (Sodium-dependent Glucose Transporter, z.B. im Darm) über Membranen transportiert (Brown, 2000). Transportproteine können auch für den Transport von pharmazeutischen Wirkstoffen über die Membran ausgenützt werden (Amidon & Sadée, 1999).
Abb. 1.4 Aktivierung des Prodrugs Oseltamivir zum aktiven Wirkstoff Oseltamivircarboxylat.
Transporter transportieren aber auch in die Gegenrichtung (sog. Efflux). Lipophile Substanzen, die in die Membran diffundiert sind, werden so wieder aus der Membran zurück in den Darm transportiert. Der am besten charakterisierte Efflux-Transporter ist P-Glykoprotein (P-gp), ein ATP-abhängiger Transporter. Pgp wurde zunächst als Faktor bei der Entstehung der Kreuzresistenz von Tumorzellen gegenüber Zytostatika entdeckt (Gottesman, Fojo & Bates, 2002). Später wurde gezeigt, dass der Transporter auch an physiologischen Geweben, z.B. im Magen-Darm-Trakt, vorkommt (Thiebaut et al., 1987). In den 90er Jahren wurde beispielsweise mit Knockout-Mäusen gezeigt, dass P-gp die Funktion einer Transportbarriere gegenüber Xenobiotika und pharmazeutischen Wirkstoffen wahrnimmt (z.B. Zhang & Benet, 1999). Als weiterer Transportweg ist noch die Rezeptor-abhängige Endo- und Transzytose zu nennen, bei dem Vesikel, die durch einen Einstülpungsvorgang der Zellmembran entstehen, den Wirkstoff einschließen und in und über die Zelle transportieren.
Die diskutierten Transportprozesse an der Zellmembran sind jedoch nicht nur bei der Absorption im Darm relevant, sondern auch bei allen weiteren pharmakokinetischen Prozessen, welchen der Wirkstoff im Organismus ausgesetzt ist, d.h. der Biotransformation, der Verteilung und der Elimination. Bei der ersten Leberpassage (hepatischer First-Pass) strömt das venöse Blut aus dem Darm an den Leberzellen (Hepatozyten) vorbei in Richtung Herz (Welsch, 2006). Neben zahlreichen Funktionen im Stoffwechsel hat die Leber durch ihre strategische Position zwischen Darm und systemischem Kreislauf eine wichtige Entgiftungs- und Ausscheidungsfunktion. Substanzen ohne physiologische Funktionen, sogenannte Xenobiotika (z.B. Zusatzstoffe in der Nahrung, Drogen oder Agrochemikalien) können hier durch Enzyme biotransformiert und der Ausscheidung in die Galle oder der Niere zugeführt werden (Testa, 1995; 2006). So werden lipophile Wirkstoffe beispielsweise besser wasserlöslich gemacht, damit sie effizient eliminiert werden können (Beyer, 1990). Durch diese erste Leberpassage kann ein wesentlicher Anteil des Wirkstoffs inaktiviert und eliminiert werden (z.B. Thummel, Kunze & Shen, 1997).
Eine kausale Bedingung für die hepatische Biotransformation (den Metabolismus) und die hepatische Elimination ist die Aufnahme in die Leberparenchymzellen (Hepatozyten) über deren sinusoidale Zellmembran, da die metabolischen Reaktionen vorwiegend intrazellulär stattfinden. Bei dieser Aufnahme sind wiederum dieselben Transportprozesse involviert, die oben bereits diskutiert wurden. Der biotransformierte und unveränderte Wirkstoff kann wieder zurück ins Blut oder über eine weitere Membran in die Galle transportiert und der Ausscheidung zugeführt werden (Transporter an der Leber: Kullak-Ublick, Stieger & Meier, 2004).
Hat der Wirkstoff nach der ersten Leberpassage schließlich die systemische Zirkulation erreicht, muss er aus dem Blut zu seiner molekularen Zielstruktur gelangen. Wenn diese außerhalb der Blutgefäße liegt, muss er dazu in das Gewebe verteilt werden (Distribution). Je nach Zielstruktur müssen erneut zelluläre Barrieren überwunden werden, zum Beispiel die Blut-Hirn-Schranke, die vom abgedichteten Endothel der Blutgefäße im Gehirn ausgebildet wird. Die Passage aus den Blutgefäßen in das Hirngewebe wird durch diese Barriere erschwert. Wiederum entscheiden die diskutierten Transportprozesse, die Eigenschaften des Wirkstoffs und die spezifischen Eigenschaften der Barriere, ob eine Substanz diese Barriere passieren kann. Die Blut-Hirn-Schranke stellt ein großes Problem für die Entwicklung zentral wirksamer Medikamente dar, da sie generell für Wirkstoffe schlecht durchlässig ist (Pardridge, 2005).
Sobald der Wirkstoff in den Organismus aufgenommen ist, beginnt auch seine Ausscheidung (Elimination). Neben der hepatischen Elimination sind insbesondere die Nieren an der Ausscheidung der Wirkstoffe beteiligt (renale Elimination). Alternative Ausscheidungswege, zum Beispiel über die Lunge, sind möglich und für einige Xenobiotika bedeutsam (z.B. Methanol). Die renale Klärung des Blutes wird von drei Prozessen bestimmt, der Filtration, der Reabsorptionund der Sekretion.
Die Reabsorption und die Sekretion sind erneut Prozesse, bei denen der Wirkstoff eine Zellschicht und Zellmembranen passieren muss (oder parazellulär transportiert wird). Wiederum sind die diskutierten Transportprozesse beteiligt und determinieren zusammen mit den Eigenschaften des Wirkstoffs (und weiteren Faktoren, z.B. der Proteinbindung) das quantitative Ausmaß der Ausscheidung.
Oseltamivir, welches nach der Aktivierung durch Hydrolyse als Oseltamivircarboxylat vorliegt, wird vorwiegend renal ausgeschieden. Es wird sowohl filtriert als auch sekretiert. Da Oseltamivircarboxylat eine negative Ladung trägt, erfolgt die Sekretion vermutlich über Organische Anionen-Transporter (OAT). Da die Sekretion mit Probenecid inhibiert werden kann (Hill et al., 2002) wurde auch spekuliert, dass im Fall einer Grippepandemie das rare Oseltamivir mit Probenecid gestreckt werden könnte (Butler, 2005). Dies analog zu Penicillin, dessen Wirkdauer durch Probenecid signifikant verlängert werden kann (z.B. Burnell & Kirby, 1951).
Zusammenfassend sind Transportprozesse und Transportbarrieren entscheidend am Schicksal eines pharmazeutischen Wirkstoffs im Organismus beteiligt, d.h. an seiner Absorption, der Distribution, am Metabolismus und an der Elimination. Diese Prozesse, die mit der Freisetzung (Liberation) unter dem Begriff der Pharmakokinetik zusammengefasst werden (Dost, 1953; 1968), stehen wiederum in direktem Zusammenhang mit der Pharmakodynamik (d.h. den Effekten des Wirkstoffs, unerwünschten Wirkungen, Interaktionen und Kontraindikationen). Schließlich sind Transportproteine auch selbst interessante Targets für die Pharmakotherapie. So gehören etwa die Protonenpumpen-Inhibitoren (z.B. Omeprazol, Antramups®, Target: H+/K+-ATPase), die Antidepressiva (z.B. Fluoxetin, Fluctine®...
Inhaltsverzeichnis
- Cover
- Series Page
- Title Page
- Copyright
- Dedication
- Vorwort
- 1: Einleitung
- 2: Freisetzung aus der Arzneiform
- 3: Gastrointestinale Absorption
- 4: Prozesse an der Leber
- 5: Verteilung zum Target
- 6: Elimination an der Niere
- Sachverzeichnis
Häufig gestellte Fragen
Ja, du kannst dein Abo jederzeit über den Tab Abo in deinen Kontoeinstellungen auf der Perlego-Website kündigen. Dein Abo bleibt bis zum Ende deines aktuellen Abrechnungszeitraums aktiv. Erfahre, wie du dein Abo kündigen kannst
Nein, Bücher können nicht als externe Dateien, z. B. PDFs, zur Verwendung außerhalb von Perlego heruntergeladen werden. Du kannst jedoch Bücher in der Perlego-App herunterladen, um sie offline auf deinem Smartphone oder Tablet zu lesen. Erfahre, wie du Bücher herunterladen kannst, um sie offline zu lesen
Perlego bietet zwei Pläne an: Essential und Complete
- Essential ist ideal für Lernende und Fachkräfte, die es genießen, eine Vielzahl von Themen zu erkunden. Greife auf die Essential Library mit über 800.000 vertrauenswürdigen Titeln und Bestsellern in den Bereichen Wirtschaft, persönliche Weiterentwicklung und Geisteswissenschaften zu. Enthält unbegrenzte Lesezeit und Standard-Vorlesestimme.
- Complete: Perfekt für fortgeschrittene Lernende und Forschende, die vollen, uneingeschränkten Zugriff benötigen. Entsperre über 1,5 Millionen Bücher zu Hunderten von Themen, einschließlich akademischen und spezialisierten Titeln. Der Complete-Plan enthält außerdem fortschrittliche Funktionen wie Premium Vorlesen und Forschungsassistent.
Wir sind ein Online-Lehrbuch-Abonnement-Service, bei dem du für weniger als den Preis eines einzelnen Buchs pro Monat Zugriff auf eine gesamte Online-Bibliothek erhältst. Bei über 1,5 Millionen Büchern zu mehr als 990 Themen bist du bestens versorgt! Erfahre mehr über unsere Mission
Achte auf das Symbol zum Vorlesen bei deinem nächsten Buch, um zu sehen, ob du es dir auch anhören kannst. Bei diesem Tool wird dir Text laut vorgelesen, wobei der Text beim Vorlesen auch grafisch hervorgehoben wird. Du kannst das Vorlesen jederzeit anhalten, beschleunigen und verlangsamen. Erfahre mehr über die Funktion „Vorlesen“
Ja! Du kannst die Perlego-App sowohl auf iOS- als auch auf Android-Geräten nutzen, damit du jederzeit und überall lesen kannst – sogar offline. Perfekt für den Weg zur Arbeit oder wenn du unterwegs bist.
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Ja, du kannst auf Moderne Pharmakokinetik von Beat Ernst,Alexander Vögtli im PDF- und/oder ePUB-Format sowie auf andere beliebte Bücher in Physical Sciences & Industrial & Technical Chemistry zugreifen. In unserem Katalog stehen über 1,5 Millionen Bücher zur Verfügung.