Mit Inkrafttreten der Medizinprodukteverordnung (EU) 2017/745 (MDR) am 25. Mai 2017 und deren Beginn der Gültigkeit nach einer Übergangszeit von drei Jahren werden neue gesetzliche Vorschriften für die Markteinführung von Medizinprodukten auf dem Markt der Europäischen Union entstehen. Die Geschwindigkeit der Entwicklung der MDR wurde 2012 durch den PIP-Implantatskandal in Frankreich beschleunigt. Die MDR zielt darauf ab, medizinische Geräte für Benutzer und Patienten sicherer und effizienter zu machen. Die neuen regulatorischen Anforderungen üben Druck auf Hersteller, benannte Stellen und Behörden gleichermaßen aus. Diese Arbeit soll die Risiken und Chancen der MDR für Hersteller aktiver Medizinprodukte der Risikoklasse IIa untersuchen. Basierend auf einer Literaturrecherche der geltenden Standards wurden die Risiken und Chancen für Hersteller aktiver Medizinprodukte der Risikoklasse IIa im Vergleich zum konventionellen Verfahren nach Richtlinie 93/42/EWG anhand der Konformitätsbewertung ermittelt. Interviews mit Experten von Behörden, benannten Stellen und Entwicklern geben Informationen über das Bewusstsein der Interessengruppen und praktische Ansätze zur Umsetzung der Anforderungen. Sowohl die Literaturrecherche als auch die Experteninterviews zeigen, dass die Anforderungen, die bereits vor der Einführung des MDR bestanden, erheblich waren und Fragen zur zukünftigen Innovationskraft deutscher und europäischer Medizintechnikunternehmen offenbleiben. Es ist jedoch bereits jetzt klar, dass die Folgen des Inkrafttretens des MDR für Klein- und Kleinstunternehmen dramatisch sein können.

eBook - ePub
Die neue Verordnung (EU) für Medizinprodukte 2017/745
Chancen und Risiken für Hersteller unter besonderer Berücksichtigung des Konformitätsbewertungsverfahrens
- 124 Seiten
- German
- ePUB (handyfreundlich)
- Über iOS und Android verfügbar
eBook - ePub
Die neue Verordnung (EU) für Medizinprodukte 2017/745
Chancen und Risiken für Hersteller unter besonderer Berücksichtigung des Konformitätsbewertungsverfahrens
Über dieses Buch
375,005 Studierende vertrauen auf uns
Zugang zu über 1,5 Millionen Titeln zu einem fairen monatlichen Preis.
Mit unseren Lerntools kannst du noch effizienter lernen.
Information
1 Einleitung
Die Medizintechnik als Teil der Gesundheitswirtschaft ist in Deutschland eine der erfolgreichsten Branchen der Industrie (Plagens, 2001, S. 3). Der Bundesverband Medizintechnologie (BVMed) führt im „Branchenbericht Medizintechnologien 2018“ hinsichtlich der Bedeutung der Gesundheitswirtschaft in Deutschland Folgendes an: „Die Gesundheitswirtschaft weist im Vergleich zur Gesamtwirtschaft überdurchschnittliche Wachstumsraten auf. Sie beschäftigt rund sieben Millionen Menschen und erwirtschaftet über 350 Milliarden Euro. Das entspricht einem Anteil von rund 11 Prozent am Bruttoinlandsprodukt. 10,2 Prozent des deutschen Außenhandelsüberschusses gehen auf die Gesundheitswirtschaft zurück.“ (BVMed 2018, S.3)
Speziell für die Branche der Medizinprodukte und Medizintechnik als Teilbereich der Gesundheitswirtschaft lässt sich festhalten: Mit einer Bruttowertschöpfung von rund 13,2 Mrd. Euro generiert die Branche im Kernbereich rund 3,9 % der gesamten Bruttowertschöpfung der Gesundheitswirtschaft (Bundesministerium für Wirtschaft und Energie, 2017, S. 2).
Die Medizintechnik ist aber nicht nur eine der erfolgreichsten Branchen in Deutschland, sie ist auch eine der am stärksten innovationsgetriebenen. Immer kürzere Produktlebenszyklen machen es notwendig, dass die Unternehmen Entwicklungsprozesse immer schneller optimieren, um Durchlaufzeiten zu verkürzen und Gewinnspannen zu erhöhen, und „[…] dies zeigt sich besonders an den hohen Investitionsaufwendungen des Wirtschaftszweigs in Forschung und Entwicklung (F&E) sowie der Anzahl an Patentanmeldungen“ (Zippel, 2016, S. 1). Entwicklungen in medizintechnischen Unternehmen beziehen sich nicht nur auf die Neuentwicklung von Apparaten und Geräten zur Diagnose sowie zur Behandlung von Menschen, sondern auch auf den Bereich der sogenannten Design-Changes, der die Änderung bzw. Verbesserung eines bestehenden Produktes umfasst.
Bei diesen Änderungen stehen in der Regel die Verbesserung sowie Anpassung an die Bedürfnisse und Anforderungen der Kunden im Vordergrund. Eine Änderung an Produkten kann jedoch beispielsweise auch durch Bauteiländerungen in Verbindung mit Kostensenkungen, Bauteilabkündigungen oder durch eine Aufrechterhaltung der Normenkonformität hervorgerufen werden. Mit wachsenden Anforderungen und dadurch entstehenden neuen Produkten werden den Anwendern von Medizintechnik neue Möglichkeiten zur Therapie von Patienten an die Hand gegeben. Umso bedeutsamer wird es für die Unternehmen, stabile Prozesse zur Inverkehrbringung von Medizinprodukten zu etablieren und Wissen aufzubauen, um die künftigen Anforderungen an die MDR (Medical Device Regulation) zu erfüllen. Für die Entwicklung und Zulassung von Medizinprodukten vergehen teilweise fünf oder mehr Jahre, je nachdem, in welchem Bereich das Produkt eingesetzt wird (Gerber, 2009, S. 2). Die Produkte der modernen Medizin werden „immer komplexer“ und „Patienten und Ärzte erwarten von den Herstellern sicherere und innovativere Medizinprodukte“ (Gerber, 2009, S. 1), die erfolgreichere und schonendere Einsätze an den Patienten ermöglichen sollen.
Die DIN EN ISO 9001 und die DIN EN ISO 13485 – letztere ist speziell bezogen auf die Medizintechnikbranche – sind Qualitätsmanagementsysteme, aus denen Forderungen hinsichtlich des Umgangs und Aufbaus von Entwicklungsprozessen hervorgehen. Normative und gesetzliche Anforderungen werden für Unternehmen im Bereich der Medizintechnik zunehmend strenger und werden zudem durch Stellen wie den TÜV oder die DEKRA in regelmäßigen Audits überprüft. Dies hat zur Folge, dass der Prozess von der Idee bis zur Inverkehrbringung eines Produktes zunehmend zeitaufwändiger und kostspieliger wird. Die Unternehmen stehen damit vor der Herausforderung, zum einen die internen Entwicklungsprozesse zu optimieren und zum anderen externes Wissen über die regulatorischen Anforderungen einzukaufen oder aufzubauen, um marktfähig und zukunftssicher agieren zu können.
Die Verordnung (EU) über Medizinprodukte 2017/745 (MDR) wurde am 25. Mai 2017 im Amtsblatt der Europäischen Union veröffentlicht. Sie gilt ab dem 26. Mai 2020 für alle Mitgliedsstaaten der Europäischen Union. In Erwägungsgrund 1 der MDR wird folgendes beschrieben: „[...] einen soliden, transparenten, berechenbaren und nachhaltigen Rechtsrahmen für Medizinprodukte zu schaffen, der ein hohes Niveau an Sicherheit und Gesundheitsschutz gewährleistet, gleichzeitig aber innovationsfördernd wirkt [...]" (Verordnung (EU) 2017/745, ErwGr. 1). Oberste Priorität in der Erwägung für die MDR hat das Schutzniveau für Patienten und Anwender. Dabei beziehen sich die Autoren im Erwägungsgrund 2 auf die in der Verordnung geforderten hohen Standards an die Qualität und Sicherheit der Medizinprodukte. Dabei werden auch kleine und mittelständische Unternehmen berücksichtigt. Es soll eine Harmonisierung für die Inverkehrbringung und die Inbetriebnahme von Medizinprodukten auf dem europäischen Binnenmarkt entstehen, ohne dabei den freien Warenverkehr negativ zu beeinflussen (Verordnung (EU) 2017/745, ErwGr. 2).
Mit dem Inkrafttreten der MDR werden die Hersteller im Bereich der Medizinprodukte einmal mehr mit neuen und höheren Anforderungen an die Inverkehrbringung von Medizinprodukten konfrontiert. Die Einführung der MDR wurde durch den sogenannten PIP-Skandal im Jahr 2012 beschleunigt. Das französische Unternehmen Poly Implant Prothèse (PIP) hatte anstelle eines Silikons, das zur Produktion von Implantaten zugelassen ist, ein Silikon in Industriequalität zur Produktion von Implantaten verwendet und diese Produkte auf den Markt gebracht (Gemke, 2017, S. 15). Die Europäische Union (EU) (2012) äußerte in einer Pressemitteilung den Bedarf zum unverzüglichen Handeln. Darin wurde der damalige EU-Kommissar für Gesundheit und Verbraucher, John Dalli, zitiert, der eine strikte und uneingeschränkte Rechtsvorschrift für Medizinprodukte forderte; die Kontrolle und die Sicherheit von Medizinprodukten müssten verschärft werden (Europäische Kommission, 2012). Dalli stellte einen ‚PIP-Action-Plan‘ auf, wonach vier wesentliche Bereiche gestärkt werden sollen:
- Funktion der benannten Stellen
- Marktüberwachung
- Koordinierung der Überwachung
- Kommunikation und Transparenz
(Europäische Kommission)
Ausgelöst durch Schadensersatzklagen von vom PIP-Skandal Betroffenen wurde erstmalig die rechtliche Relevanz eines zertifizierten Qualitätsmanagementsystems diskutiert. Dabei ging es um den Stellenwert eines solchen Systems in Fragen der europäischen Produkthaftung (Oeben, 2016, S. 39).
Derzeit müssen Medizinprodukte ein Konformitätsbewertungsverfahren (KBV) zur Conformité Européene(CE)-Kennzeichnung durchlaufen, bevor sie in Verkehr gebracht werden können. Dessen Ablauf wird bis zum Inkrafttreten der MDR durch die Medical-Device-Directive (MDD) geregelt. Letztere ist dabei die englische Bezeichnung für die RL (Richtlinie) 93/42/EWG. Ab dem Jahr 2020 wird das Verfahren zur CE-Kennzeichnung durch die MDR vorgegeben. Dies wird alle Marktbeteiligten, v. a. jedoch die Medizinprodukte-Industrie, vor erhebliche Schwierigkeiten stellen, weil die neuen Anforderungen auch für Medizinprodukte gelten, die bereits seit Jahrzehnten unverändert in Verkehr gebracht werden (Graf, 2017, S. 963). Mit der Veröffentlichung der MDR im Amtsblatt der Europäischen Union am 05.05.2017 ergeben sich folgende Umsetzungsfristen für die Medizintechnikunternehmen:
- 25.05.2017 Inkrafttreten der neuen Verordnung
- 26.05.2020 Geltungsbeginn der Verordnung
- 26.05.2024 spätester Ablauf der Zertifikate nach MDD, die nach dem 26.04.2017 ausgestellt worden sind
- 27.05.2025 Ablauf der ‚Abverkaufsregelung‘: letztmöglicher Tag der ‚Bereitstellung‘ oder ‚Inbetriebnahme‘ von Produkten. Vorausaussetzung ist jedoch, dass diese nach altem Recht zuvor legal in Verkehr gebracht worden sind (Hill, 2017, S. 110). Diese Situation könnte zum Tragen kommen, wenn sich die Medizinprodukte bereits physisch beim Endkunden befinden und dort eingelagert sind.
Hieraus resultierend müssen die Hersteller jedoch ab dem 26. Mai 2020 die Anforderungen der Medizinprodukte-Verordnung (EU) erfüllen, um Medizinprodukte weiter in Verkehr bringen zu dürfen.
1.1 Zielsetzung der Arbeit und Forschungsfragen
Die vorliegende Arbeit setzt sich mit der Frage nach den Folgen der MDR für die Hersteller von Medizinprodukten in Deutschland auseinander. Die zentrale Forschungsfrage lautet:
Welche Chancen und Risiken ergeben sich für Hersteller von aktiven Medizinprodukten der Risikoklasse IIa voraussichtlich durch die Einführung und Umsetzung der neuen Medizinprodukte-Verordnung (EU), d. h. der MDR?
Im Rahmen dieser generellen und zentralen Fragestellung soll besonders danach gefragt werden,
- wie sich Prozesse der Inverkehrbringung von Medizinprodukten durch die Einführung der MDR voraussichtlich verändern und
- welchen Einfluss die Einführung der MDR voraussichtlich auf zukünftige Entwicklungen und Innovationen in der Medizintechnik in Deutschland haben wird.
Hierbei ist zu berücksichtigen, dass die Arbeit im Vorfeld der verbindlichen Anwendung der MDR erstellt wurde und Aussagen sowie Erfahrungswerte von Unternehmen über die tatsächliche Anwendung der MDR den benannten Stellen sowie den Behörden noch nicht vorlagen.
Aufgrund der Vielzahl und großen Variation der Medizinprodukte, die gemäß EU-Verordnung (Verordnung (EU) 2017/745, Kap. V, Abschn. 1, Art. 51, Abs. 1) und unter Verwendung von insgesamt 22 Klassifizierungsregeln (gemäß Verordnung (EU) 2017/745, Anhang VIII, Kapitel I) ihrem Risiko nach in drei Klassen – I, IIa, IIb und III – unterteilt sind, sowie einer entsprechend großen Variation der Anforderungen zur legalen Inverkehrbringung von medizintechnischen Geräten konnten im Rahmen der vorliegenden Arbeit nicht alle Medizinprodukte berücksichtigt werden. Daher wurde die Betrachtung auf Medizinprodukte der Klasse IIa, auf die die meisten der aktiven Medizinprodukte entfallen, beschränkt (vgl. Kapitel 2).
1.2 Aufbau der Arbeit und Vorgehensweise bei der Beantwortung der Forschungsfrage
Die Arbeit besteht aus zwei Teilen: Der erste umfasst die Kapitel 2 und 3. In Kapitel 2 werden die rechtlichen Rahmenbedingungen der Inverkehrbringung von Medizinprodukten beschrieben. In Kapitel 3 werden die Entwicklung, das Qualitätsmanagement und das KBV von Medizinprodukten erläutert. Dieser Teil der Arbeit basiert auf einer Literaturrecherche, die mit Hilfe von Suchbegriffen (Keywords) durchgeführt wurde, von denen der Autor der vorliegenden Arbeit aufgrund seines bis dahin bestehenden Vorwissens vermutete, dass sie dabei helfen würden, eine strukturierte Analyse der thematisch relevanten, vorliegenden Literatur vorzunehmen. Diese Suchbegriffe sind in der folgenden Mindmap dargestellt:
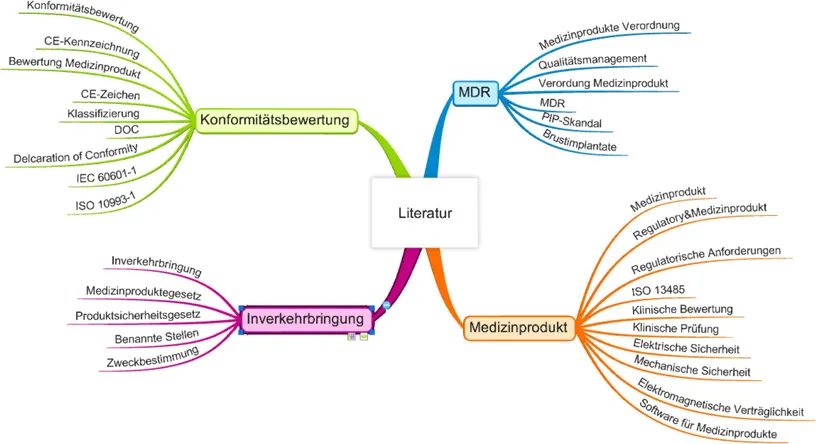
Abbildung 1 – Mindmap der Suchbegriffe für Literaturrecherche, eigene Darstellung
Nach der persönlichen Bewertung der Keywords durch den Autor sind diese in die eigentliche Recherche eingeflossen. Für die Recherche über die Suchbegriffe wurden die Bool’schen Operatoren AND, OR und NOT verwendet (Läzer et al., 2010, S. 6).
Der zweite Teil der Arbeit umfasst eine empirische Untersuchung, mit der Antworten auf die oben genannten Fragen aufgrund von Experteninterviews gegeben werden. Genauer gesagt sollen die Experteninterviews die Literaturanalyse ergänzen und das Bild, das sich aufgrund der Literaturanalyse von der zukünftig erwartbaren Entwicklung in der Medizintechnik ergibt, vervollständigen. Ihnen ist Kapitel 4 der vorliegenden Arbeit gewidmet. Für die Durchführung von Experteninterviews spricht, dass aufgrund der Aktualität der neuen Gesetzgebung für Medizinprodukte in Form der MDR einerseits noch keine in standardisierter Form abfragbaren Erfahrungswerte vorliegen und andererseits vorhandenes Expertenwissen nur teilweise erhoben bzw. publiziert ist. Die Auswertung der Interviews erfolgte anhand einer an Mayring (2008) angelehnten qualitativen Inhaltsanalyse.
Das inhaltliche Schlusskapitel der vorliegenden Arbeit bildet Abschnitt 5. In ihm werden die Ergebnisse der Experteninterviews zu jenen der Literaturanalyse ins Verhältnis gesetzt und auf der Basis dieses Vergleichs wird die in dieser Einleitung formulierte Forschungsfrage hinsichtlich der erwartbaren Folgen des Inkrafttretens der MDR für die Hersteller von Medizinprodukten beantwortet.
2 Rechtliche Rahmenbedingungen der Inverkehrbringung von Medizinprodukten
Der rechtliche Rahmen der Inverkehrbringung von Medizinprodukten wird gesteckt durch Verordnungen, Regelungen, Normen und Gesetze, die bestimmen, wie Hersteller, Inverkehrbringer sowie benannte Stellen bei der Entwicklung, Verbreitung und Prüfung von Medizinprodukten zu verfahren haben. Deren Verständnis – auch das der MDR – ist nur möglich, wenn die Definitionen zentraler Begriffe gekannt werden, auf die sie sich beziehen. In diesem Kapitel werden die wichtigsten Verordnungen, Regelungen, Normen und Gesetze für die Inverkehrbringung von Medizinprodukten kurz vorgestellt. Anschließend wird erläutert, wie in ihnen definiert ist, was als Medizinprodukt angesehen wird, wer als Hersteller bzw. wer als Inverkehrbringer von Medizinprodukten gilt und wer sogenannte ,benannte Stellen‘ sind, die prüfen, ob Medizinprodukte den Sicherheitsanforderungen genügen.
2.1 Richtlinien, Verordnungen, Normen und Gesetze
In Verordnungen, Richtlinien, nationalen Gesetzen und technischen Normen sind die Anforderungen formuliert, die die Hersteller für die legale Inverkehrbringung von Produkten in der Europäischen Union erfüllen müssen. Dadurch soll sichergestellt werden, dass in sämtlichen EU-Mitgliedsstaaten dieselben Anforderungen an und Voraussetzungen für die Produktion von Medizinprodukten für die Hersteller bestehen. Auf dieser Basis soll damit der freie Verkehr von Waren mit gleichermaßen hohem Sicherheitsniveau innerhalb des europäischen Binnenmarktes gesichert werden (Krey & Kapoor, 2017, S. 165).
Die Europäische Kommission (2016) hat in einer Bekanntmachung einen Leitfaden für die Umsetzung der Produktvorschriften der EU, den sog. ‚Blue Guide‘, herausgegeben. Durch diesen sollen die Harmonisierungen der Rechtsvorschriften in Bezug auf die wesentlichen Anforderungen an Produkte auf dem EU-Markt geregelt werden. Festgehalten wurde darin u. a., dass technische Spezifikationen für Produkte in harmonisierten Normen geregelt sein sollen, sofern diese den wesentlichen Anforderungen einer Rechtsvorschrift, etwa einer Richtlichtlinie oder Verordnung, entsprechen. Wenn ein Produkt nach anzuwendenden harmonisierten Normen hergestellt und in Verkehr gebracht wird, kann damit angenommen werden, dass hierdurch die Rechtsvorschriften durch den Hersteller eingehalten werden. Gestärkt werden soll zudem die Anwendung von Qualitätsmanagementsystemen der ISO-9001-Reihe, die in der Europäischen Union harmonisiert wurden (Euro...
Inhaltsverzeichnis
- Abstract
- Inhaltsverzeichnis
- 1. Einleitung
- 2. Rechtliche Rahmenbedingungen der Inverkehrbringung von Medizinprodukten
- 3. Literaturanalyse
- 4. Empirische Forschung
- 5. Zusammenfassung, Schlussfolgerungen und Ausblick
- 6. Literaturverzeichnis
- 7. Abbildungsverzeichnis
- 8. Tabellenverzeichnis
- 9. Abkürzungsverzeichnis
- 10. Anhang
- Impressum
Häufig gestellte Fragen
Ja, du kannst dein Abo jederzeit über den Tab Abo in deinen Kontoeinstellungen auf der Perlego-Website kündigen. Dein Abo bleibt bis zum Ende deines aktuellen Abrechnungszeitraums aktiv. Erfahre, wie du dein Abo kündigen kannst
Nein, Bücher können nicht als externe Dateien, z. B. PDFs, zur Verwendung außerhalb von Perlego heruntergeladen werden. Du kannst jedoch Bücher in der Perlego-App herunterladen, um sie offline auf deinem Smartphone oder Tablet zu lesen. Erfahre, wie du Bücher herunterladen kannst, um sie offline zu lesen
Wir sind ein Online-Lehrbuch-Abonnement-Service, bei dem du für weniger als den Preis eines einzelnen Buchs pro Monat Zugriff auf eine gesamte Online-Bibliothek erhältst. Bei über 1,5 Millionen Büchern zu mehr als 990 Themen bist du bestens versorgt! Erfahre mehr über unsere Mission
Achte auf das Symbol zum Vorlesen bei deinem nächsten Buch, um zu sehen, ob du es dir auch anhören kannst. Bei diesem Tool wird dir Text laut vorgelesen, wobei der Text beim Vorlesen auch grafisch hervorgehoben wird. Du kannst das Vorlesen jederzeit anhalten, beschleunigen und verlangsamen. Erfahre mehr über die Funktion „Vorlesen“
Ja! Du kannst die Perlego-App sowohl auf iOS- als auch auf Android-Geräten nutzen, damit du jederzeit und überall lesen kannst – sogar offline. Perfekt für den Weg zur Arbeit oder wenn du unterwegs bist.
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Ja, du kannst auf Die neue Verordnung (EU) für Medizinprodukte 2017/745 von Patrick Walitschek im PDF- und/oder ePUB-Format sowie auf andere beliebte Bücher in Technik & Maschinenbau & Maschinenbau Allgemein zugreifen. In unserem Katalog stehen über 1,5 Millionen Bücher zur Verfügung.