There has been a fundamental change in our understanding of multiple sclerosis in recent years, affecting the pathogenesis of the condition as well its treatment, diagnosis and care. This volume presents these advances in a clear and application-oriented way and reflects all areas of the current state of knowledge on multiple sclerosis. As a reference work as well as a guide, it provides specific recommendations on diagnosis and therapy, with a focus on practical issues and aspects of patient care. In addition to background information about suspected pathological mechanisms, approaches to treatment monitoring are presented using diagrams. The book is rounded off by an overview of proven and new drugs, their mechanisms of action and aspects that need to be taken into account when there is a possible change of therapy. The book represents a unique companion for everyday clinical practice in the German-speaking countries.
375,005 Studierende vertrauen auf uns
Zugang zu über 1 Million Titeln zu einem fairen monatlichen Preis.
Mit unseren Lerntools kannst du noch effizienter lernen.
Die Multiple Sklerose (MS) ist eine chronisch entzündliche Erkrankung des zentralen Nervensystems (ZNS), die sich meist im frühen Erwachsenenalter manifestiert (Dendrou et al. 2015; Gilhus et al. 2016). Nach wie vor stellt sie die häufigste neurologische Ursache frühzeitiger Behinderung im jungen Erwachsenenalter dar. Die Erkrankung weist eine große klinische, bildgebende und pathologische Heterogenität auf und wird in verschiedene Verlaufs- und Sonderformen unterteilt. Die individuelle Prognose wird durch ein komplexes Zusammenspiel von genetischer Prädisposition, Umweltfaktoren und spezifischer Erregerexposition bestimmt, wobei die Details noch nicht abschließend geklärt sind (Wiendl und Kieseier 2010).
Bei der Mehrzahl der Patienten beginnt die Erkrankung mit einem schubförmig remittierenden Verlauf (RRMS = relapsing-remitting multiple sclerosis), bei dem sich Phasen der klinischen Verschlechterung, Remission und Stabilität ablösen. Als Erstmanifestation der schubförmigen MS wird das klinisch isolierte Syndrom angesehen, bei dem die Patienten bei klinisch, MR-tomografisch und labormedizinisch begründetem Verdacht noch nicht alle Kriterien zur Diagnose einer schubförmigen MS erfüllen. In späteren Krankheitsphasen wird der schubförmige Verlauf bei einem Teil der Patienten von einer sekundären Progression (kontinuierliche Zunahme der neurologischen Defizite) abgelöst (SPMS). Bei 10–20 % der Patienten beginnt die Erkrankung mit einer stetigen (primär progredienten) neurologischen Verschlechterung (PPMS), die mit oder ohne aufgelagerte Schübe abläuft. Die MS kann alle zentralnervösen Strukturen betreffen, daher kommt es bei den Schüben sowie im Verlauf der Erkrankung zu Symptomen wie Sensibilitätsstörungen, Lähmungen, Seh- und Koordinationsstörungen, kognitiven Einschränkungen, psychiatrischen Auffälligkeiten, Müdigkeit (Fatigue), Schmerzen und Störungen bzw. Ausfall der Blasenkontrolle. Die Symptome variieren im Verlauf und sind interindividuell sehr unterschiedlich. Im Rahmen eines Schubes entwickeln sich die neurologischen Symptome über mehrere Tage, erreichen dann ein Plateau und bilden sich über Tage bis Wochen zurück (Schumacher et al. 1965). Die Remissionstendenz sowie das Ansprechen auf die Akuttherapie des Schubes sind zu Beginn der Erkrankung im Allgemeinen besser als im weiteren Krankheitsverlauf.
Als Schub werden abzugrenzende neu aufgetretene klinische Symptome angesehen, die subjektiv berichtet oder durch objektive Untersuchung verifiziert werden können und länger als 24 Stunden andauern (Poser et al. 1983; McDonald et al. 2001). Unterschieden davon werden Pseudoattacken oder Pseudoexazerbationen, also paroxysmale Verschlechterungen durch Änderungen der Körperkerntemperatur (Uhthoff-Phänomen; Uhthoff 1890) oder im Rahmen von Infekten. Der Verdacht auf stattgehabte Schübe kann sich aus subjektiven Patientenberichten und der Anamnese ergeben. Für die Diagnosestellung einer MS sind allerdings objektivierbare klinische Befunde korrespondierend zu einer entsprechenden ZNS-Läsion erforderlich. Einzelne paroxysmale Episoden (wie z. B. tonische Spasmen) werden nicht als Schub definiert. Multiple derartige Episoden mit einer Dauer von mehr als 24 Stunden werden jedoch ebenfalls als Schub gewertet. Das Kriterium für das Vorliegen zweier separater Schübe ist definitionsgemäß ein Zeitintervall von 30 Tagen zwischen dem Beginn des ersten und des zweiten Schubes (Poser et al. 1983; McDonald et al. 2001; Polman et al. 2011; Polman et al. 2005). Schubveränderungen oder neue Schübe innerhalb von 30 Tagen oder weniger bezeichnet man als Schubkomplex.
1.2 Verlauf und Prognose
Die schubförmige MS ist eine Erkrankung des jungen Erwachsenenalters und manifestiert sich meist zwischen dem 20. und 40. Lebensjahr (Median 28 Jahre). Nur wenige Patienten entwickeln die Erkrankung vor dem 15. oder nach dem 55. Lebensjahr, wenn auch die Häufigkeit der Diagnose Multiple Sklerose im Kindesalter zunimmt (
Kap. 1.3.1). Im klinischen Gebrauch wird die Definition von Lublin aus dem Jahr 2013 verwendet; die im Vergleich zur früheren Einteilung insbesondere das klinisch isolierte Syndrom als Frühstadium mit aufnahm (Lublin und Reingold 1996; Lublin et al. 2014). In den Kriterien wurde das klinisch isolierte Syndrom als mögliche Erstmanifestation einer Multiplen Sklerose gewertet, bei dem das Kriterium der zeitlichen Dissemination jedoch noch nicht erfüllt ist (
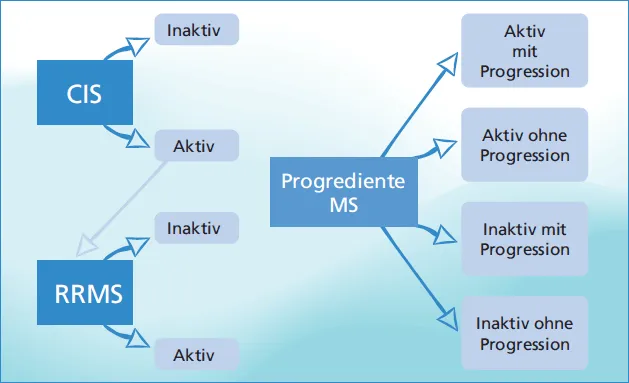
Abb. 1.1 Verlaufsformen der MS). Die Anerkennung des klinischen isolierten Syndroms (CIS) ist für die frühe Therapie und Prognose von Bedeutung. Das radiologisch isolierte Syndrom wurde dagegen nicht als neue Verlaufsform gewertet.
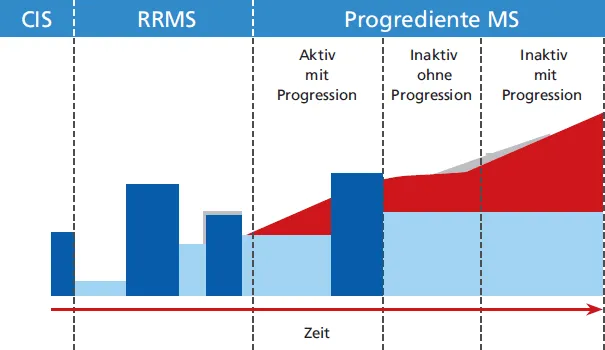
Weiterhin werden verschiedene Formen der progredienten MS unterschieden, bei der es schubunabhängig zu einer Zunahme der Behinderung kommt. Dabei handelt es sich um die sekundär chronisch progrediente Form (SPMS), bei der es nach einem zunächst schubförmigen Verlauf zu einer schleichenden schubunabängigen Verschlechterung kommt, und die primär chronisch progrediente Form (PPMS) ohne Schübe. Es existieren bislang keine im Alltag gebräuchlichen Kriterien, die die Transition von einer RRMS in eine SP-MS definieren. 2016 wurden Kriterien vorgeschlagen, die zur Definition das Fortschreiten der Behinderung ohne Schübe, bestätigt nach drei Monaten, einen EDSS ≥ 4 und einen Pyramidenbahnscore ≥ 2 umfassen (Lorscheider et al. 2016). Die progredienten Verlaufsformen der SPMS (sekundär progredient) und der PPMS (primär progredient) werden zusammen als PMS bezeichnet. In den 2013 definierten Kriterien wurden als zusätzliche Parameter die Aktivität und Progression einer Erkrankung eingeführt, wobei die Aktivität die klinischen Schübe und die MR-tomografische Krankheitsaktivität umfasst und die Progression das Fortschreiten klinischer Behinderung bei den progredienten Verlaufsformen (Lublin et al. 2014). Dieses soll insbesondere in Therapiestudien eine bessere Vergleichbarkeit des Patientenkollektivs gewährleisten. Die Begriffe einer benignen oder malignen Multiplen Sklerose sollten dagegen nicht mehr verwendet werden, da die Beurteilung aufgrund von Schwankungen im individuellen Verlauf nur retrospektiv erfolgen kann.
Bei der Mehrzahl der Patienten (etwa 85 %) manifestiert sich die Erkrankung mit einem schubförmig remittierenden Verlauf (RRMS)
Abb. 1.1a: Verlaufsformen: Einordnung der Verlaufsformen (2013), adaptiert nach Lublin et al. (2014) Aktivität bestimmt anhand klinischer Schübe und/oder MRT-Aktivität (Kontrastmittel aufnehmende Läsionen; neue oder eindeutig vergrößerte T2-Läsionen). Progression bei mindestens jährlicher Untersuchung); bei nicht verfügbaren Untersuchungsergebnissen: Aktivität »unbestimmt«. Die Progression wird durch eine klinische Beurteilung gemessen, die mindestens einmal jährlich durchgeführt wird. Wenn keine Beurteilungen verfügbar sind, sind Aktivität und Progression »unbestimmt«. CIS: klinisch isoliertes Syndrom(geht in schubförmig-remittierende MS (RRMS) über, sobald anhand weiterer Krankheitsaktivität die aktuellen Diagnosekriterien der MS erfüllt). PPMS: primär progrediente MS; PRMS: progredient remittierende MS; SPMS: sekundär progrediente MS. PP: fortschreitende Akkumulation von Behinderung ab Krankheitsbeginn, SP: fortschreitende Akkumulation von Behinderung nach zunächst schubförmigem Verlauf.
Abb. 1.1b: Aktuelle Einordnung der Verlaufsformen (eigene Abbildung)
(Weinshenker et al. 1989). Die Erkrankten haben Schübe bzw. Episoden akuter neurologischer Dysfunktionen, die sich häufig innerhalb weniger Wochen wieder zurückbilden. Die Erstsymptome der MS unterscheiden sich bei Patienten mit frühem und späterem Erkrankungsalter. Bei jüngeren Patienten beginnt die RRMS häufig monosymptomatisch mit einer Optikusneuritis (36 %) oder Parästhesien (33 %). Paresen allein oder in Kombination mit sensiblen Ausfällen findet man häufiger bei älteren Patienten (50 %). Neue neurologische Dysfunktionen entwickeln sich typischerweise über mehrere Stunden bis wenige Tage. Paroxysmal auftretende Symptome wie neuropathische oder neuralgiforme Schmerzen oder dystone Bewegungen kommen bei der MS ebenfalls häufig vor. Zur Definition des Schubs siehe Kapitel 4.4.1. Ein Schub dauert meist ein bis drei Wochen, selten länger als acht Wochen, wobei letzterer eine schlechtere Rückbildungstendenz aufweist. Ob es zu einer vollständigen Remission kommt, hängt jedoch nicht nur von der Schubdauer, sondern auch von den jeweiligen Krankheitszeichen ab. Parästhesien, Optikusneuritis oder Doppelbilder bilden sich zumindest zu Beginn der Erkrankung meistens gut zurück. Paresen, zerebelläre Symptome oder autonome Störungen haben dagegen eine schlechtere Prognose. Obwohl einige Patienten im gesamten Krankheitsverlauf jeweils komplett remittierende Schübe haben, wird in späteren Krankheitsphasen der schubförmige Verlauf bei einem Teil der Patienten von einer sekundären Progression (kontinuierliche Zunahme der neurologischen Defizite) abgelöst (SPMS), die mit oder ohne zusätzliche Schübe abläuft. Die Rückbildung der Schübe ist dann zumeist inkomplett, zudem ist eine schleichende Progression der Behinderung feststellbar. Das Risiko, nach der Diagnose eines klinisch isolierten Syndroms innerhalb von fünf Jahren eine klinisch sichere Multiple Sklerose zu entwickeln, liegt – abhängig von den MRT-Befunden zum Diagnosezeitpunkt und der Präsenz oligoklonaler Banden – zwischen 30 und 87 % (Kuhle et al. 2015). Patienten, bei denen zu Beginn der Erkrankung oligoklonale Banden (OKB) nachgewiesen werden können, haben ein mehr als doppelt so hohes Risiko wie Patienten ohne OKB-Nachweis im Liquor. Ebenso ist ein erhöhter Immunglobulin (Ig)G-Index mit einer höheren Konversionsrate assoziiert. Patienten mit zwei bis neun Läsionen im MRT zu Krankheitsbeginn haben ein mehr als doppelt so hohes Risiko, eine MS zu entwickeln als Patienten mit keiner oder nur einer Läsion. Das Risiko eines erneuten Schubes steigt darüber hinaus mit der Zahl der Läsionen an und der Abstand zwischen den Schüben nimmt statistisch ab. Allerdings haben auch Patienten mit sehr wenigen Läsionen zu Beginn der Erkrankung ein hohes Risiko, in den ersten zehn Jahren nach dem CIS eine klinisch sichere MS zu entwickeln (Simon et al. 2015; Voortman et al. 2017; van der Vuurst de Vries et al. 2017), sodass in Zukunft möglicherweise noch weitere Marker zur Risikoeinschätzung gefunden werden müssen. Der frühzeitige Einsatz von Immuntherapien verzögert die Konversion zu einer klinisch definitiven MS signifikant und reduziert vermutlich die Behinderungsprogression, zumindest die schubgebundene (Spelman et al. 2016; Jokubaitis et al. 2015; Kappos et al. 2016; Comi et al. 2009). Nach einer isolierten Optikusneuritis liegt das Risiko, eine klinisch sichere Multiple Sklerose zu entwickeln, mit etwa 50 % etwas niedriger. Möglicherweise ist dieses auch einer Unschärfe bei der Diagnose der Optikusneuritiden geschuldet. Eine Übersicht dazu findet sich bei Miller et al. 2005a, Soderstrom 2001, Miller et al. 2005a und b und Sorensen et al. 1999. Auch bei der Optikusneuritis sind der Nachweis oligoklonaler Banden sowie eine höhere Läsionslast im MRT prädiktive Faktoren (Soderstrom et al. 1998; Skov et al. 2011; Miller et al. 2005a; Miller et al. 2005b; Tintore et al. 2006; Swanton et al. 2010).
Während der ersten zwei Jahre nach Diagnosestellung hat ein Großteil aller Patienten mit einem schubförmigen Verlauf eine klinische und paraklinische Krankheitsaktivität. Zumindest in der Frühphase der RRMS werden Schubfrequenzen von 0,5–1,3/Jahr beobachtet. Zu den nachgewiesenen, den Schub provozierenden Faktoren gehören virale Infekte (Sibley et al. 1985). Obwohl bakterielle Infekte per se offenbar das Risiko für Schübe nicht steigern, können Infektionen als solche (z. B. Harnwegsinfekt) eine signifikante symptomatische Verschlechterung bewirken, die bei der klinischen Evaluation in Betracht gezogen werden muss. Eine Beziehung zwischen einer Verschlechterung oder Schüben und Stress, Traumata und chirurgischen Eingriffen ist in verschiedenen anekdotischen Berichten zu finden. Bis heute allerdings fehlen überzeugende Beweise für solche Assoziationen (Goldacre et al. 2006; D’Hooghe et al. 2010). Sobald die sekundär progrediente Verlaufsform erreicht ist, steigt das Risiko bleibender Behinderungen. Während in früheren Studien ohne Medikamente von einer Konversion in eine sekundär-progrediente Form nach im Median zehn Jahren ausgegangen wurde, zeigt eine neuere Kohortenstudie, dass nach zehn Jahren nur 6,4 % der Patienten eine sekundär-progrediente Verlaufsform erreicht haben, nach 20 Jahren waren es 24,2 % (University of California et al. 2016). In den früheren Untersuchungen vor dem Einsatz von Immuntherapien, benötigten Patienten im Median nach 15 Jahren eine einseitige Gehhilfe, die neueren Untersuchungen zeigen einen deutlich langsameren Anstieg der Behinderung. Nach im Median 16,8 Jahren erreichten nur 10,7 % einen EDSS ≥ 6 (Weinshenker et al. 1989 a, b; University of California et al. 2016). Wenngleich die langsamere Progression im Vergleich zu früheren Untersuchungen nicht beweisend auf die zu Verfügung stehenden Immuntherapien zurückgeführt werden kann, zeigen Untersuchungen, dass Medikamente gegen eine aktive/hochaktive Verlaufsform die Behinderungsprogression verzögern, und zwar auch dann, wenn bereits eine moderate Behinderung bestand (Lizak et al. 2017). Das Europäische Komitee für Behandlung und Erforschung der Multiple Sklerose (ECTRIMS) und die Europäische Akademie für Neurologie (EAN) haben sich 2016 in einer gemeinsamen Stellungnahme für einen frühzeitigen Therapiebeginn bei CIS und einen frühzeitigen Wechsel zu einer wirksameren Therapie ausgesprochen (ECTRIMS 2016), sodass bei einer weiteren konsequenten Frühtherapie eine weitere Verbesserung der Prognose erwartet wird.
Merke
Der frühzeitige Einsatz von Immuntherapien verzögert die Konversion zu einer klinisch definitiven MS signifikant und reduziert ebenfalls die schubgebundene Be...
Inhaltsverzeichnis
Deckblatt
Titelseite
Impressum
Abkürzungsverzeichnis
Inhalt
Vorwort
Danksagung
1 Klinisches Bild und Verlauf
2 Epidemiologie und Genetik
3 Pathogenese
4 Diagnose
5 Therapie
6 Patientenrelevanter Informationsteil
Literatur
Stichwortverzeichnis
Häufig gestellte Fragen
Ja, du kannst dein Abo jederzeit über den Tab Abo in deinen Kontoeinstellungen auf der Perlego-Website kündigen. Dein Abo bleibt bis zum Ende deines aktuellen Abrechnungszeitraums aktiv. Erfahre, wie du dein Abo kündigen kannst
Nein, Bücher können nicht als externe Dateien, z. B. PDFs, zur Verwendung außerhalb von Perlego heruntergeladen werden. Du kannst jedoch Bücher in der Perlego-App herunterladen, um sie offline auf deinem Smartphone oder Tablet zu lesen. Erfahre, wie du Bücher herunterladen kannst, um sie offline zu lesen
Perlego bietet zwei Abopläne an: Elementar und Erweitert
Elementar ist ideal für Lernende und Profis, die sich mit einer Vielzahl von Themen beschäftigen möchten. Erhalte Zugang zur Basic-Bibliothek mit über 800.000 vertrauenswürdigen Titeln und Bestsellern in den Bereichen Wirtschaft, persönliche Weiterentwicklung und Geisteswissenschaften. Enthält unbegrenzte Lesezeit und die Standardstimme für die Funktion „Vorlesen“.
Pro: Perfekt für fortgeschrittene Lernende und Forscher, die einen vollständigen, uneingeschränkten Zugang benötigen. Schalte über 1,4 Millionen Bücher zu Hunderten von Themen frei, darunter akademische und hochspezialisierte Titel. Das Pro-Abo umfasst auch erweiterte Funktionen wie Premium-Vorlesen und den Recherche-Assistenten.
Beide Abopläne sind mit monatlichen, halbjährlichen oder jährlichen Abrechnungszyklen verfügbar.
Wir sind ein Online-Lehrbuch-Abo, bei dem du für weniger als den Preis eines einzelnen Buches pro Monat Zugang zu einer ganzen Online-Bibliothek erhältst. Mit über 1 Million Büchern zu über 990 verschiedenen Themen haben wir bestimmt alles, was du brauchst! Erfahre mehr über unsere Mission
Achte auf das Symbol zum Vorlesen bei deinem nächsten Buch, um zu sehen, ob du es dir auch anhören kannst. Bei diesem Tool wird dir Text laut vorgelesen, wobei der Text beim Vorlesen auch grafisch hervorgehoben wird. Du kannst das Vorlesen jederzeit anhalten, beschleunigen und verlangsamen. Erfahre mehr über die Funktion „Vorlesen“
Ja! Du kannst die Perlego-App sowohl auf iOS- als auch auf Android-Geräten nutzen, damit du jederzeit und überall lesen kannst – sogar offline. Perfekt für den Weg zur Arbeit oder wenn du unterwegs bist. Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Ja, du hast Zugang zu Multiple Sklerose von Heinz Wiendl,Catharina Korsukewitz,Bernd C. Kieseier im PDF- und/oder ePub-Format sowie zu anderen beliebten Büchern aus Medicine & Neurology. Aus unserem Katalog stehen dir über 1 Million Bücher zur Verfügung.