![]()
Apoptosis and Angioneogenesis
Christiansen H, Christiansen NM (eds): Progressive Neuroblastoma: Innovation and Novel Therapeutic Strategies.
Pediatr Adolesc Med. Basel, Karger, 2015, vol 20, pp 59-80 (DOI: 10.1159/000382087)
______________________
Neuroblastoma and the p53 Pathway
Lindi Chen · Deborah A. Tweddle
Newcastle Cancer Centre, Northern Institute for Cancer Research, Newcastle University, Newcastle, UK
______________________
Abstract
The tumor suppressor protein p53 is a transcription factor which plays a crucial role in maintaining genomic stability through the regulation of a vast repertoire of downstream target genes involved in cellular processes, such as cell cycle arrest, apoptosis, senescence, differentiation and DNA repair. Under normal cellular conditions, p53 is tightly regulated by its critical negative regulator MDM2, an E3 ligase which targets p53 for ubiquitin-mediated degradation. p14ARF is a tumor suppressor protein and the negative regulator of MDM2. Together, p53, MDM2 and p14ARF form an autoregulatory loop frequently inactivated in many human cancers. MDM2-p53 antagonists are small-molecule inhibitors designed to disrupt the interaction between p53 and MDM2 thereby nongenotoxically activating p53 in tumors with wild-type p53. Several MDM2-p53 antagonists are undergoing early clinical evaluation in adults and are anticipated to enter pediatric trials in the near future. In contrast to several other cancers, neuroblastoma is a predominantly p53 wild-type tumor; however, p53 pathway inactivation through MDM2 amplification and p14ARF aberrations have been reported. Nongenotoxic activation of wild-type p53 using MDM2-p53 antagonists alone and in combination with other agents offers a novel therapeutic strategy for patients with neuroblastoma to potentially improve survival and/or reduce the toxicity associated with current chemotherapy regimens.
© 2015 S. Karger AG, Basel
p53
The p53 tumor suppressor protein, encoded by the TP53 gene located at 17p13.1, is a 53-kDa nuclear phosphoprotein consisting of an acidic N-terminal transactivation domain, a proline (Pro)-rich domain, a central sequence-specific DNA-binding domain, a tetramerization domain and a highly basic C-terminal regulatory domain. Nuclear export signals are located within both the N- and C-terminus, and three lysine-rich nuclear localization signals are located within the C-terminal. p53 functions as a transcription factor playing a crucial role in maintaining genomic stability through the regulation of a vast repertoire of downstream target genes involved in cellular processes, including but not limited to cell cycle arrest, apoptosis, senescence, differentiation and DNA repair [1]. Identified more recently, p53 family members p63 and p73 share structural and functional homology with p53. It is now known that several p53, p63 and p73 isoforms exist and can be abnormally expressed in human cancer [2].
The p53/MDM2/p14ARF Pathway
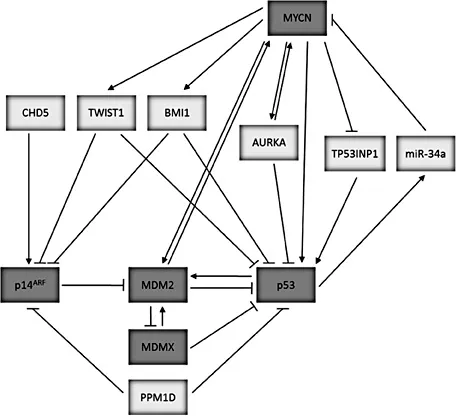
Under normal cellular conditions, p53 is tightly regulated by MDM2, an E3 ubiquitin ligase that is a direct transcriptional target of p53 and induced in response to p53 activation. MDM2 can bind directly to p53 to inhibit p53 transcriptional activity, promote nuclear export and target p53 for ubiquitin-mediated proteasome degradation. This critical regulation of p53 by MDM2 is supported by the observed embryonic lethality of MDM2 knockout mice and their rescue by the concomitant deletion of p53. p14ARF is a tumor suppressor protein which promotes p53 stability and activity by directly binding and inhibiting MDM2. The latter can lead to inhibition of MDM2 E3 ligase activity and p53 nuclear export, sequestration of MDM2 within the nucleolus, and promotion of MDM2 degradation. Activated p53 can subsequently downregulate the expression of p14ARF, so that p53, MDM2 and p14ARF form an autoregulatory loop. MDMX is a homologue of MDM2, which does not possess intrinsic E3 ligase activity but can inhibit p53 activity through direct binding and by enhancing MDM2-mediated ubiquitination of p53 (fig. 1; reviewed by Chen and Tweddle [3]).
p53 Pathway Inactivation and Cancer
Since its identification in 1979, the importance of p53 in tumor suppression has been reaffirmed by reports that TP53 is the most frequently mutated gene in human cancer - up to half of all patients with cancer. This is further supported by the observed predisposition to early-onset cancer, and in very rare cases neuroblastoma, in individuals with Li-Fraumeni syndrome and the development of a range of spontaneous tumors in p53 null murine models, most commonly T-cell lymphomas.
In addition to inactivating TP53 mutations, the p53 pathway can also be abrogated by overexpression or amplification of MDM2 or MDMX, and methylation or deletion of p14ARF. Similar to p53-null mice, spontaneous tumor development in mice with MDM2 or MDMX overexpression, or deficient p14ARF has been observed, albeit at a slower rate. Aberrations of TP53, MDM2, MDMX or p14ARF tend to be mutually exclusive; however, it is not clear why certain mechanisms of p53 pathway inactivation are favored in some tumors but not others, and is most likely influenced by the various selective pressures acting upon the cancer (reviewed by Chen and Tweddle [3]).
The frequency of TP53 mutations varies from 10 to 70% across different cancer types, being more common in solid tumors compared with hematological malignancies and in adult tumors compared with pediatric cancers [4]. The majority of mutations are missense mutations and occur within the DNA-binding domain, in particular at 6 mutational hotspots: R175, G245, R248, R249, R273 and R282 [1]. A smaller proportion of mutations detected are frameshift or nonsense mutations. TP53 mutations identified to date can be found within publically available p53 databases such as the International Agency for Research on Cancer (IARC) TP53 Mutation Database (http://www-p53.iarc.fr/) and The p53 Website (http://p53.free.fr/). In some cases, TP53 mutational inactivation is accompanied by loss of heterozygosity at the TP53 loci. Interestingly, in contrast to TP53, TP63 and TP73 are rarely mutated in human cancers and neither p63 nor p73 knockout mice exhibit an increased susceptibility to developing spontaneous tumors (reviewed by Chen and Tweddle [3]).
Fig. 1. The p53/MDM2/p14ARF pathway and potential mechanisms of inactivation in neuroblastoma.
Owing to the requirement for p53 to function as an active tetramer, TP53 mutations therefore exert a dominant negative effect. The majority of p53 mutants are nonfunctional, while a small proportion retain partial p53 wild-type (wt) transcriptional function. It is now known that p53 mutants possess oncogenic p53 wt independent gain-of-functions, which has been most significantly demonstrated in murine models of p53 mutants. Broadly speaking, the gain-of-function attributes of p53 mutants are due to mutant p53-mediated transcription of novel target genes and/or interaction with cellular proteins [1].
The prognostic and predictive significance of TP53 somatic mutations in human cancers have been extensively evaluated and studies have demonstrated associations between p53 mutations and poor prognosis and/or therapy resistance in several cancers, such as colorectal and breast cancer. In addition to mutations, there are also several single nucleotide polymorphisms (SNPs) affecting the p53 pathway which can influence cancer risk and clinical outcome, including TP53 codon 72, MDM2 SNP309 and p21 codon 31.
p53 and Neuroblastoma
In contrast to many other human cancers, neuroblastoma is a predominantly TP53 wt tumor where mutations have been reported to occur in ~3% of cases analyzed to date (reviewed by Chen and Tweddle [3]). Initial studies observed an accumulation of p53 in neuroblastoma, which may be attributed to the embryonic nature of the disease, reflecting a failure of precursor cells to mature. In support of the latter, retinoic acid-induced differentiation of neuroblastoma cells has been accompanied by a decrease in p53 expression, and siRNA-mediated p53 inhibition in neuroblastoma cells has resulted in a more morphologically differentiated phenotype (reviewed by Chen and Tweddle [3]). Early studies suggested that p53 was sequestered within the cytoplasm and nonfunctional, and several mechanisms have been described, including p53 C-terminal nuclear localization signal masking, hyperactive nuclear export, binding to the cytoplasmic anchor, Parc, aberrant p53 hyperubiquitination and MDMX/MDM2-mediated cytoplasmic tethering (reviewed by Chen and Tweddle [3]). However, most studies (including our own) have demonstrated nuclear and functional p53. Despite this, abnormalities in the p53 pathway have been shown to be involved in neuroblastoma pathogenesis, progression, relapse and drug resistance, and will be discussed below.
p53 Pathway Polymorphisms in Neuroblastoma
Polymorphisms in components of the p53 pathway can influence cancer risk and clinical outcome, and have been shown for several cancers. A SNP at codon 72 in exon 4 of the TP53 gene results in an arginine (Arg) to Pro substitution (rs1042522). In the context of wt p53, studies have observed that p53-72R is more effective at inducing apoptosis in comparison with p53-72P, whereas in the context of mutant p53, p53-72R can bind with greater affinity and inhibit the proapoptotic functions of p73 compared with p53-72P. A study of TP53 codon 72 variations in 286 newly diagnosed neuroblastomas observed that the Pro/Pro genotype wa...