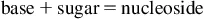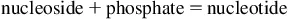All cell and tissue functions are ultimately governed by gene expression. Consequently, the reasons for electing to study the modulation of RNA levels as at least one parameter of the cellular biochemistry may be as diverse as the intracellular RNA population itself. Generally speaking, the characterization of RNA is almost always related to transcription, i.e., gene expression questions being asked in the context of a particular scientific inquiry, and most often revolves around measuring the dynamic abundance level of one or several transcripts.
The goals in any experimental design involving RNA generally revolve around one or more fundamental themes, including but not limited to the following:
1. Measurement of the steady-state abundance of cellular transcripts. Steady-state RNA refers to the net accumulation of transcription products in the cell, or in a subcellular compartment such as the nucleus or the cytoplasm. It is the combined result of RNA synthesis, stability, and degradation. This is the most common reason why RNA is isolated from cells and tissues. Analysis may focus on one transcript, a few transcripts, or all transcripts simultaneously; this latter approach is commonly known as global analysis of gene expression or whole transcriptome profiling. Given the ease with which RNA can be purified from biological sources, the use of various sensitive, contemporary approaches is widespread for generating quantitative and qualitative profiles of RNA populations using any of a variety of laboratory techniques.
2. Synthesis of complementary DNA (cDNA). Unstable, single-stranded messenger RNA (mRNA) can serve as the template for the in vitro synthesis of very stable single- or double-stranded cDNA molecules. This is the first step for subsequent amplification by the polymerase chain reaction (PCR), often for some “quantitative purpose,” for transcript mapping purposes, for direct ligation into a vector for sequencing or for expression of the encoded protein, for the physical separation of two or more cDNA species, or for the older strategy of synthesizing an entire cDNA library (older literature occasionally refers to a cDNA library as a “clone bank.”) which can be propagated for long-term storage and analysis. In any event, the construction of cDNA is the creation of a permanent biochemical record of the cell at the moment of cellular disruption. Historically, the synthesis of highly representative cDNA is one of the most important methodologies in the molecular biology laboratory and, in some hands, remains a significant challenge.
3. Detection of viruses which harbor an RNA genome. This proceeds via the synthesis of cDNA, as described above, followed by PCR or another cDNA amplification method.
4. Identification of the transcription start site (TSS). Historically, mapping of RNA molecules, including the 5′ end, the 3′ end, and the size and location of introns, was accomplished via nuclease protection assay, as described in Chapter 18, Quantification of Specific mRNAs by Nuclease Protection. Now, however, transcript mapping is now almost always performed by some variant of 5′- or 3′-rapid amplification of cDNA ends (RACE; see Chapter 8: RT-PCR: A Science and an Art Form). As it is well known that a single genetic locus often has the potential to produce multiple RNAs, each with a different TSS and often in a tissue-specific manner, TSS mapping is an invaluable technique.
5. Measurement of the rate of transcription of gene sequences or the pathways of RNA processing. This may be deduced, at least in part, by the nuclear run-on assay in which radiolabeled ribonucleotide precursors are incorporated into nascent transcripts in direct proportion to the abundance of each species of RNA being transcribed (see Chapter 19: Analysis of Nuclear RNA). When used in conjunction with other methods that examine steady-state RNA levels, the regulation of genes can often be assigned as transcriptional or due to posttranscriptional events.
6. In vitro translation of purified mRNA. The resulting polypeptide may be further characterized by immunoprecipitation or Western analysis. Cell-free translation represents an older method for the identification of specific transcripts: by providing the raw materials needed to support translation, one is able to demonstrate that a transcript of putative identity is able to support the synthesis of the cognate peptide. For example, this approach could be used to demonstrate that two transcripts from the same genetic locus with alternative TSSs are, in fact, able to direct the synthesis of identical or closely related proteins. In applications such as rational drug design, in vitro translation is helpful because understanding the three-dimensional architecture of a protein, and its wild type or mutated function(s), may suggest novel applications in the area of functional proteomics.