![]()
Alford RL, Sutton VR (eds): Medical Genetics in the Clinical Practice of ORL.
Adv Otorhinolaryngol. Basel, Karger, 2011, vol 70, pp 84-90
______________________
Multiple Endocrine Neoplasia: Types 1 and 2
Deborah J.Marsha · Oliver Gimmb,c
aHormones and Cancer Group, Kolling Institute of Medical Research, Royal North Shore Hospital, University of Sydney E25, St. Leonards, N.S.W., Australia; bDepartment of Surgery and cInstitute for Clinical and Experimental Medicine (IKE), University Hospital, Linköping, Sweden
______________________
Abstract
Multiple endocrine neoplasia type 1 (MEN 1) and type 2 (MEN 2) are autosomal-dominantly inherited syndromes where highly penetrant germline mutations predispose patients to the development of tumours in hormone-secreting cells. In the case of MEN 1, loss-of-function germline mutations in the tumour suppressor gene MEN1 increase the risk of developing pituitary, parathyroid and pancreatic islet tumours, and less commonly thymic carcinoids, lipomas and benign adrenocortical tumours. In the case of MEN 2, gain-of-function germline mutations clustered in specific codons of the RET proto-oncogene increase the risk of developing medullary thyroid carcinoma (MTC), phaeochromocytoma and parathyroid tumours. Offering RET testing is best practice for the clinical management of patients at-risk of MEN 2, and MEN 2 has become a classic model for the integration of molecular medicine into patient care. Prophylactic thyroidectomy in an asymptomatic RET mutation carrier to address the risk of developing MTC can prevent or cure this malignancy. No similar preventative strategies can be employed to prevent or cure MEN 1-associated tumours. Genetic testing for MEN 1 is therefore both more complex due to a general lack of mutational hotspots, and the benefit to patients is less straight forward. While a number of genotype-phenotype correlations exist in MEN 2, providing further rationale for performing genetic testing in this condition, these correlations are absent in MEN 1. This review summarises our current knowledge of these two syndromes with emphasis on those aspects with specific relevance to the otorhinolaryngologist.
Copyright © 2011 S. Karger AG, Basel
Multiple endocrine neoplasia (MEN) types 1 and 2 are part of a group of rare familial endocrine neoplasia disorders characterised by the presence of tumours in hormone secreting cells along with other disorders including von Hippel-Lindau disease, familial paraganglioma and phaeochromocytoma, Cowden syndrome, hyperparathyroidism jaw-tumour syndrome and Carney complex (reviewed in Marsh and Zori [1]). MEN 1 and MEN 2 fall at either end of the spectrum when considering both the ease of conducting genetic testing, and the ability to act on genetic information to prevent or cure a tumour.
Multiple Endocrine Neoplasia Type 1
Clinical Presentation/Phenotype
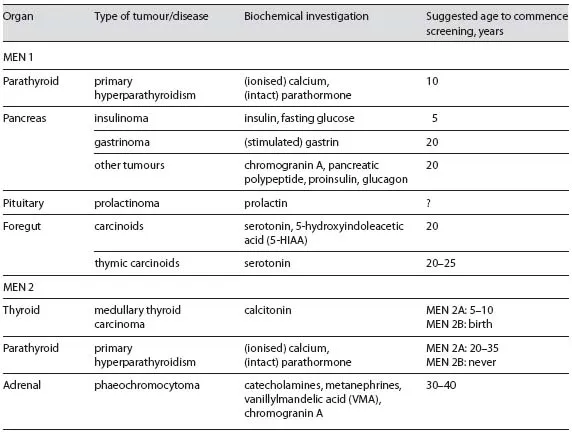
MEN 1 has been estimated to occur in between 1 in 10,000 to 1 in 100,000 people [2] and is characterised by neuroendocrine tumours of the gastro-enteric-pancreatic tract. These lesions include parathyroid tumours causing primary hyperparathyroidism (pHPT), pancreatic islet cell tumours, anterior pituitary tumours, and less commonly thymic carcinoids, lipomas, thyroid tumours, benign tumours of the adrenal cortex, and atypically generally benign tumours of the adrenal medulla (phaeochromocytoma); however, presentation can vary markedly between two individuals (table 1)
Table 1. Clinical presentations of patients with MEN 1 and MEN 2
Diagnostic criteria for MEN 1 specifies that an individual must have abnormalities in at least two of the more commonly affected endocrine glands, as well as a first-degree relative with at least one MEN 1-related lesion or a known MEN1 mutation [3]. Almost 50% of MEN 1 patients will succumb to MEN 1-related disease, most frequently associated with pancreatic islet cell tumours [4]. Almost all patients with MEN 1 develop pHPT that can lead to hypercalcemia throughout their lifetime, and although this is almost always benign, its recurring nature can make surgical management difficult. In comparison to sporadic pHPT, these patients typically present between 20 and 25 years of age, approximately 30 years earlier than their sporadic counterparts [5]. Biochemical screening is recommended from the age of 10 years. The underlying genetic cause implies that all parathyroid glands can be affected. In fact, by the time of diagnosis, most patients already present with multiple parathyroid gland enlargement classified as either parathyroid hyperplasia or multiple adenomas. In MEN 1 patients, the diagnosis of pHPT is made if both parathyroid hormone (PTH) and ionised calcium levels are elevated [6].
Genetic Background/Genotype
Germline mutations in the tumour suppressor gene MEN1 were first identified in 1997 in patients with MEN 1 [7, 8]. MEN1 mutations are also found rarely in the related condition Familial Isolated Hyperparathyroidism (FIH). MEN1 consists of 10 exons, where exon 1 is not translated. It encodes the 610 amino acid (67 kDa) ubiquitously expressed and predominantly nuclear protein menin that has roles in the regulation of transcription, controlling genome stability, the regulation of cell division and cell cycle control. Up to 90% of MEN 1 patients will have a MEN1 mutation, with approximately 10% of these mutations appearing to be de novo, i.e. identified in patients with apparently sporadic parathyroid adenomas [9].
Mutations are predicted to be loss of function and include small (and less frequently large) deletions, small insertions, insertion-deletions, as well as nonsense and missense point mutations and splicing mutations, with the majority predicted to prematurely truncate menin [10, 11]. There are over 570 mutation entries for MEN1 in The Human Genome Mutation Database, Cardiff, UK (http://www.hgmd.cf.ac.uk/ac/index.php), and well over 1,000 mutations reported in total [11], with mutations covering the entirety of the gene. There is some indication of potential mutational hot-spots at codons 83-84, 516 and 210-211 accounting for approximately 12% of all mutations [11]; however, in general, lack of frequent mutational clusters complicates genetic screening for this disorder. Of note, in MEN 1 patients without MEN1 mutation, germline mutations in one of the cyclin-dependent kinase inhibitor genes have been reported; however, these would appear to be rare [12].
Genotype-Phenotype Correlations
No appreciable genotype-phenotype correlations have been identified in MEN 1 [11, 13, 14].
Genetic Screening
Knowledge of a patient's MEN1 mutation carrier status is useful for clinical management strategies and life-planning decisions, but cannot avoid or cure the malignancies associated with this condition [3]. This is largely because there are no options for prophylactic surgery for MEN 1 patients, who are generally not treated until their condition first manifests. Biochemical screening to search for early disease in asymptomatic mutation carriers is recommended. If a member of a family tests negative for a MEN1 mutation that is present in affected family members, this removes them from the lifelong burden of biochemical and radiological screening for associated tumours. Guidelines have been published outlining recommended diagnostic tests and therapy for MEN1 mutation carriers [5, 10].
Surgery and Experimental Therapies
The indication to operate is given in any MEN 1 patient with the diagnosis of pHPT. Prior to primary surgery, sophisticated imaging techniques are generally not recommended since their sensitivity is low in MEN 1-associated pHPT [15]. Due to the fact that MEN 1-associated pHPT is a multiglandular disease, an attempt must be made to identify all parathyroid glands intraoperatively. This implies routine removal of the thyrothymic ligament where parathyroid glands can be found in up to 20% of patients. Once all parathyroid glands are localised, the smallest gland should be identified. A remnant about half the size of a normal gland should either be left in situ (referred to as subtotal parathyroidectomy) or be autotransplanted (referred to as total parathyroidectomy and autotransplantation). The other parathyroid glands shall be removed completely. As recurrence of the preserved tissue is common, if subtotal parathyroidectomy is performed the remnant should be marked with a non-absorbable suture or clip in order to facilitate reoperation [15].
Calcimimetics, calcium-sensing receptor agonists, show promise for the treatment of sporadic pHPT and may have a role in treating elevated PTH in the context of MEN 1 [5, 16, 17]. Depot long-acting octreotide has also been used to treat MEN 1-associated pHPT [18]. Multicentre, randomised clinical trials still need to be conducted in order to assess the efficacy of these experimental therapies.
Multiple Endocrine Neoplasia Type 2
Clinical Presentation/Phenotype
MEN 2 is believed to occur in 1 in 200,000 live births [19]. The clinical presentation of MEN 2 may vary significantly between two individuals and includes pHPT, medullary thyroid carcinoma (MTC; a malignant tumour of parafollicular thyroid C cells that secrete calcitonin) and phaeochromocytoma (phaeo; tumour of the adrenal medulla) (table 1). If two or more of these tumours are present in 1 patient or in a close relative, a diagnosis of MEN 2 should be considered. Around two thirds of people harbouring a RET mutation will develop one or more of these tumours by 70 years of age [20].
Clinically and genetically, two major types can be distinguished, namely MEN 2A and MEN 2B. In contrast to MEN 2A, patients with MEN 2B present with a Marfanoid habitus, mucosal neuromas (which often cause the lips to appear large and patulous) and ganglioneuromatosis of the gastrointestinal tract that are clues for thi...