
eBook - ePub
Pharmacovigilance Medical Writing
A Good Practice Guide
Justina Orleans-Lindsay
This is a test
Partager le livre
- English
- ePUB (adapté aux mobiles)
- Disponible sur iOS et Android
eBook - ePub
Pharmacovigilance Medical Writing
A Good Practice Guide
Justina Orleans-Lindsay
Détails du livre
Aperçu du livre
Table des matières
Citations
À propos de ce livre
Pharmacovigilance Medical Writing covers the preparation of pharmacovigilance documents for all stages of the drug development process (i.e. from clinical development through to applications for marketing authorisations to the post-marketing stage). For each document, the book presents a review of the regulatory framework that governs the content of the document, followed by practical guidance (e.g. scheduling, source data, department/functions involved in document preparation/review, appropriate timelines and planning activities), ending with a generic model document compliant with the current guidelines, which can be modified to meet specific company and product requirements.
Foire aux questions
Comment puis-je résilier mon abonnement ?
Il vous suffit de vous rendre dans la section compte dans paramètres et de cliquer sur « Résilier l’abonnement ». C’est aussi simple que cela ! Une fois que vous aurez résilié votre abonnement, il restera actif pour le reste de la période pour laquelle vous avez payé. Découvrez-en plus ici.
Puis-je / comment puis-je télécharger des livres ?
Pour le moment, tous nos livres en format ePub adaptés aux mobiles peuvent être téléchargés via l’application. La plupart de nos PDF sont également disponibles en téléchargement et les autres seront téléchargeables très prochainement. Découvrez-en plus ici.
Quelle est la différence entre les formules tarifaires ?
Les deux abonnements vous donnent un accès complet à la bibliothèque et à toutes les fonctionnalités de Perlego. Les seules différences sont les tarifs ainsi que la période d’abonnement : avec l’abonnement annuel, vous économiserez environ 30 % par rapport à 12 mois d’abonnement mensuel.
Qu’est-ce que Perlego ?
Nous sommes un service d’abonnement à des ouvrages universitaires en ligne, où vous pouvez accéder à toute une bibliothèque pour un prix inférieur à celui d’un seul livre par mois. Avec plus d’un million de livres sur plus de 1 000 sujets, nous avons ce qu’il vous faut ! Découvrez-en plus ici.
Prenez-vous en charge la synthèse vocale ?
Recherchez le symbole Écouter sur votre prochain livre pour voir si vous pouvez l’écouter. L’outil Écouter lit le texte à haute voix pour vous, en surlignant le passage qui est en cours de lecture. Vous pouvez le mettre sur pause, l’accélérer ou le ralentir. Découvrez-en plus ici.
Est-ce que Pharmacovigilance Medical Writing est un PDF/ePUB en ligne ?
Oui, vous pouvez accéder à Pharmacovigilance Medical Writing par Justina Orleans-Lindsay en format PDF et/ou ePUB ainsi qu’à d’autres livres populaires dans Medicine et Pharmacology. Nous disposons de plus d’un million d’ouvrages à découvrir dans notre catalogue.
Informations
Chapter 1
Pharmacovigilance Medical Writing – An Overview Across the Drug Development Process
A misconception considers that pharmacovigilance medical writing is concerned solely (or primarily) with the preparation of Periodic Safety Update Reports (PSURs) in the post-marketing phase of a product's life cycle. In truth, pharmacovigilance medical writing impacts on the clinical development and post-marketing phases, as well as making a significant contribution to the mandated submission documents required before the regulating authorities can grant marketing authorization/approval.
To fully appreciate the significance of pharmacovigilance medical writing within the drug development process, it is useful to take a step back and review each stage of the process and the accompanying pharmacovigilance or safety documentation. To this end, a summary outline of the key stages of the clinical development process and associated pharmacovigilance documents is presented in Figure 1.1.
Figure 1.1 Pharmacovigilance medical writing across the drug development process.
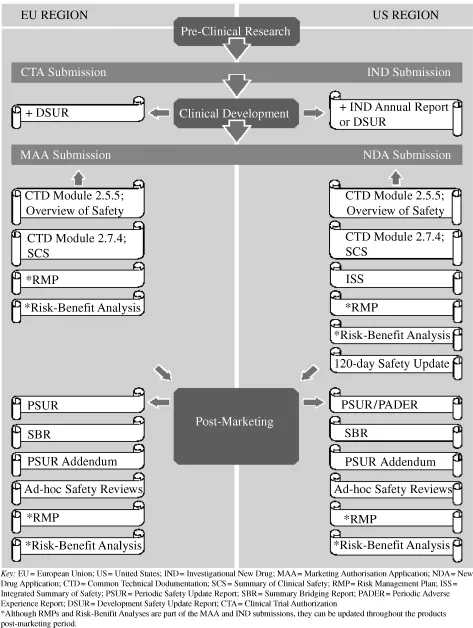
In the first instance, the clinical development phase is associated with annual submissions of the Development Safety Update Report (DSUR) in the European Union (EU) and the Investigational New Drug (IND) Annual Report in the United States (US), with submission of the DSUR also being acceptable in the US. These documents represent a mechanism, through which the safety of subjects participating in clinical studies can be monitored by the sponsoring company and the regulatory authorities, as well as ethics committees and institutional review boards.
At the time of marketing authorization applications, pharmacovigilance documents represent a significant proportion of documents contained in the submitted dossiers, including:
- Common Technical Documentation (CTD) Module 2.5.5 – Overview of Safety;
- CTD Module 2.7.4 – Summary of Clinical Safety;
- Integrated Summary of Safety (ISS);
- 120-Day Safety Update Report;
- Risk Management Plan (RMP);
- Benefit-Risk Evaluation Report.
The CTD modules (i.e. CTD Modules 2.5.5 and 2.7.4) and the ISS represent integrated analyses of all safety data collected in the clinical development of the given medicinal product, and form the basis for the product's labeling and totality of safety information that is made available to prescribers and other healthcare professionals once the product has received marketing authorization (i.e. licensed for use).
The RMP is required at the time of application for marketing authorization of most medicinal products in the EU. This document describes the safety information yet to be determined for the given medicinal product and specifies the measures that will be taken by the company to address these gaps in the product's safety profile. In addition, the RMP outlines the processes that will be taken by the company to minimize the product's known safety issues and how these efforts will be evaluated and monitored for effectiveness.
The Benefit-Risk Evaluation Report assesses the benefit derived from use of the medicinal product against the risks for a particular patient population and treated indication, to determine whether the product has a favorable benefit-risk profile (i.e. that the benefits outweigh or justify the potential risks).
After successful application for marketing authorization, a number of other pharmacovigilance documents come into effect, including:
- PSURs (or Periodic Adverse Experience Reports [PADERs] for the US region);
- PSUR Addendums;
- Summary Bridging Reports (SBR);
- RMPs and Benefit-Risk Evaluation Reports;
- Ad-hoc safety reviews.
The PSUR, PADER, and associated documents (i.e. the PSUR Addendum and SBR) are mandated for submission at periodic intervals after marketing authorization, and are intended as a means through which the Marketing Authorization Holder (MAH), that is the company granted permission to market the medicinal product, can continue to review and update the regulating authorities of the product's safety profile, so that any changes (and potential risks) can be quickly identified and addressed.
Although RMPs and Benefit-Risk Evaluation Reports are an integral part of the documents submitted for marketing authorizations, these documents will continue to be amended and updated throughout the product's post-marketing life. A number of scenarios exist that require updating of RMPs and Benefit-Risk Evaluation Reports, including:
- license renewals;
- identification of a new safety concerns;
- registration of new and clinically dissimilar indications;
- registration of treatment in a special treatment population (e.g. paediatrics and the elderly).
To afford greater utility, a separate chapter within this practitioner's manual is devoted to each phase of the drug development process that is impacted by pharmacovigilance medical writing, with a discussion of all associated pharmacovigilance or safety documents.
For ease of use and reference, the review of each pharmacovigilance document in this practitioner's manual is set out according to the following sections:
- review of regulatory requirements that underpin the preparation of each document;
- the scheduling/submission frequency for each document;
- the required data and data sources;
- the interdisciplinary team involved in the preparation and review of each document;
- an example timeline for document preparation and finalization;
- a generic model document.
The format of templates for these documents will clearly vary among different companies; however, the generic model presented for each document should provide a resource that can be modified based on therapeutic area and data requirements.
Chapter 2
Pharmacovigilance Medical Writing for Clinical Trials
2.1 Introduction
When a company or academic institution is granted permission to test a yet to be authorized medicinal product on human subjects (or an authorized medicinal product in a new patient population/indication), the sponsor of the said clinical study undertakes a legally binding obligation to provide annually to the regulatory authority, an aggregated analysis of all serious adverse drug reactions (SARs), as well as serious adverse events (SAEs) and events leading to subject withdrawal in the US, recorded from the clinical study. This is in addition to standard reporting of individual reactions in accordance with the mandated timelines. The requirement for submission of these annual reports continues until completion of the clinical studies, and is intended as an opportunity for the clinical study sponsor, ethics committees, or institutional review boards, and regulatory authorities to review and monitor the safety of subjects participating in the clinical studies.
Up until August 2011, these documents, their content, and purpose differed between the EU and US regions, with submission of the EU Annual Safety Report (ASR) and US Investigational New Drug (IND) Annual Report, respectively. The EU ASR served as an annual benefit-risk assessment exercise, and thus differed from the US IND Annual Report, which essentially functioned as an annual progress report to the Food and Drug Administration (FDA). A comparative summary of the EU ASR and US IND Annual Report is presented in Table 2.1.
Table 2.1 The EU ASR and US IND Annual Report.
| EU ASR | US IND Annual Report |
| Functions as benefit-risk assessment | Functions as progress report of the clinical program |
| Only SARs included in analysis | Includes SAEs, AEs leading to study withdrawal, and expedited safety reports |
| Covers all EU-based clinical studies and clinical studies undertaken by an EU sponsor in non-EU countries | Only covers US-based clinical studies |
| AE = adverse event; ASR = Annual Safety Report; EU = European Union; SAE = serious adverse event; SAR = serious adverse reaction; US = United States; IND = Investigational New Drug | |
However, it is no secret, that pharmacovigilance medical writing during clinical development is currently undergoing a period of transition. The EU ASR and US IND Annual Report have both been replaced by the Development Safety Update Report (DSUR), a single harmonized document that integrates both jurisdictional requirements for annual reporting of clinical trial safety data. This removes the duplication of reports to be prepared by multinational companies simultaneously sponsoring clinical studies for the same medicinal product in both re...