
eBook - ePub
Pharmaceutical Microbiological Quality Assurance and Control
Practical Guide for Non-Sterile Manufacturing
David Roesti, Marcel Goverde, David Roesti, Marcel Goverde
This is a test
- English
- ePUB (adapté aux mobiles)
- Disponible sur iOS et Android
eBook - ePub
Pharmaceutical Microbiological Quality Assurance and Control
Practical Guide for Non-Sterile Manufacturing
David Roesti, Marcel Goverde, David Roesti, Marcel Goverde
Détails du livre
Aperçu du livre
Table des matières
Citations
À propos de ce livre
Relying on practical examples from the authors' experience, this book provides a thorough and modern approach to controlling and monitoring microbial contaminations during the manufacturing of non-sterile pharmaceuticals.
- Offers a comprehensive guidance for non-sterile pharmaceuticals microbiological QA/QC
- Presents the latest developments in both regulatory expectations and technical advancements
- Provides guidance on statistical tools for risk assessment and trending of microbiological data
- Describes strategy and practical examples from the authors' experience in globalized pharmaceutical companies and expert networks
Foire aux questions
Comment puis-je résilier mon abonnement ?
Il vous suffit de vous rendre dans la section compte dans paramètres et de cliquer sur « Résilier l’abonnement ». C’est aussi simple que cela ! Une fois que vous aurez résilié votre abonnement, il restera actif pour le reste de la période pour laquelle vous avez payé. Découvrez-en plus ici.
Puis-je / comment puis-je télécharger des livres ?
Pour le moment, tous nos livres en format ePub adaptés aux mobiles peuvent être téléchargés via l’application. La plupart de nos PDF sont également disponibles en téléchargement et les autres seront téléchargeables très prochainement. Découvrez-en plus ici.
Quelle est la différence entre les formules tarifaires ?
Les deux abonnements vous donnent un accès complet à la bibliothèque et à toutes les fonctionnalités de Perlego. Les seules différences sont les tarifs ainsi que la période d’abonnement : avec l’abonnement annuel, vous économiserez environ 30 % par rapport à 12 mois d’abonnement mensuel.
Qu’est-ce que Perlego ?
Nous sommes un service d’abonnement à des ouvrages universitaires en ligne, où vous pouvez accéder à toute une bibliothèque pour un prix inférieur à celui d’un seul livre par mois. Avec plus d’un million de livres sur plus de 1 000 sujets, nous avons ce qu’il vous faut ! Découvrez-en plus ici.
Prenez-vous en charge la synthèse vocale ?
Recherchez le symbole Écouter sur votre prochain livre pour voir si vous pouvez l’écouter. L’outil Écouter lit le texte à haute voix pour vous, en surlignant le passage qui est en cours de lecture. Vous pouvez le mettre sur pause, l’accélérer ou le ralentir. Découvrez-en plus ici.
Est-ce que Pharmaceutical Microbiological Quality Assurance and Control est un PDF/ePUB en ligne ?
Oui, vous pouvez accéder à Pharmaceutical Microbiological Quality Assurance and Control par David Roesti, Marcel Goverde, David Roesti, Marcel Goverde en format PDF et/ou ePUB ainsi qu’à d’autres livres populaires dans Technology & Engineering et Quality Control in Engineering. Nous disposons de plus d’un million d’ouvrages à découvrir dans notre catalogue.
Informations
1
Microbiological Control Strategy
David Roesti1 and Marcel Goverde2
1 Novartis Pharma Stein AG, Stein, Switzerland
2 MGP Consulting GmbH, Binningen, Switzerland
CONTENTS
- 1.1 Introduction
- 1.2 Overview of a Microbial Control Strategy Program
- 1.3 Main Factors to Be Controlled
- 1.3.1 Controlled Facilities
- 1.3.2 Controlled Procedures
- 1.3.3 Controlled Product Ingredients
- 1.3.4 Controlled Utilities
- 1.3.5 Controlled Equipment
- 1.3.6 Controlled Formulation
- 1.4 Conclusion
- Bibliography
1.1 Introduction
Microbiological controls in non‐sterile pharmaceutical drug product manufacturing consist of preventing microorganisms from contaminating the final product and keeping their numbers low during the manufacturing process. By controlling the overall bioburden level, the probability of product contamination with an objectionable microorganism is also reduced. The effectiveness on the controls can be continuously evaluated with sound microbiological monitoring and trending of results.
Microbial controls can be defined with the support of risk management tools helping to find the right balance between excessive controls and contamination risk (Figure 1.1).
Figure 1.1 Balance between excessive and insufficient microbial controls.
The United States Pharmacopeia (USP) chapter <1115> provides the most recent and comprehensive guidance on factors affecting microbial controls and suggests a risk management approach to establish these controls. USP <1115> writes that microbiological risk should be assessed on a case‐by‐case basis during the development of a new product and should be evaluated during the validation of the manufacturing process.
The present chapter will likewise present high‐level multiple microbial controls that can be introduced during production of non‐sterile drug products. Details of the controls are further addressed in the corresponding chapters of this book.
A search of the FDA database for recalls (Enforcement Reports) for the category “drug products” using the keywords “microbiology,” “microbiological,” and “microbial” found 14 recalls for non‐sterile drug products (since the 1st of January 2014). Of these recalls, 10 were due to out‐of‐specification (OOS) results for microbiological specifications or aerobic microbiological count, two were due to product released to market prior to microbiological testing, and two omitted testing for a specified microorganism (source FDA website, last visited on 14 December 2018). Further details on product recalls for non‐sterile products between the years 1998 and 2006 Jimenez (2007), and 2004 and 2011 Sutton and Jimenez (2012).
1.2 Overview of a Microbial Control Strategy Program
A comprehensive microbial control program should allow to identify the risk of contamination to the product as well as the different mitigation steps to control this risk. It should also be based on the latest regulatory guidelines as well as industry best practices or current scientific knowledge and would inspire the design of facilities, equipment selection, choice of raw materials, cleaning and manufacturing procedures, etc. (Figure 1.2).
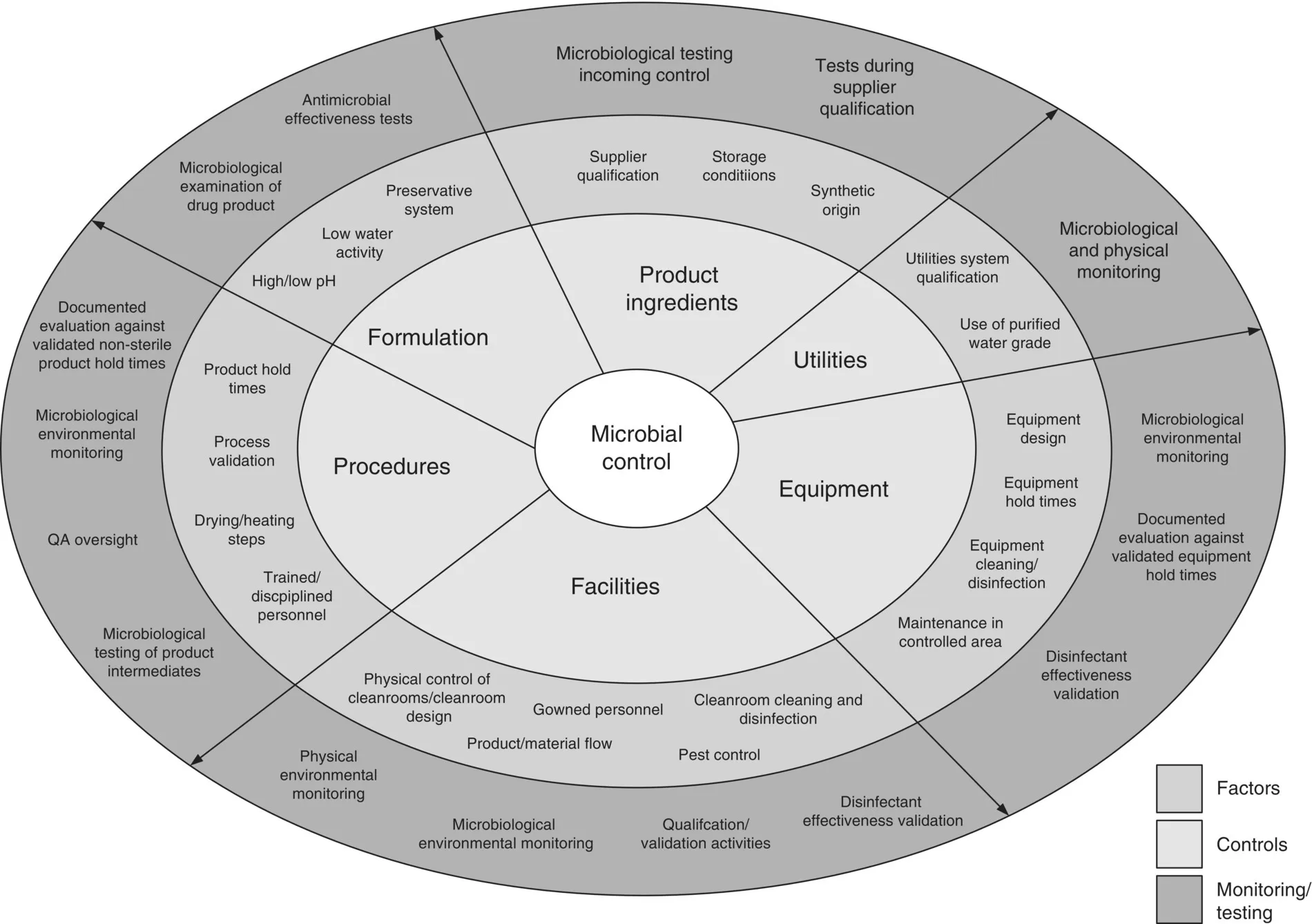
Figure 1.2 Examples of factors, controls, and monitoring implicated in a comprehensive microbial control program for non‐sterile products.
A formalized risk assessment should initially define the contamination risks and control points. A risk‐based approach is described in Chapter 2. The resulting control points and monitoring strategy serve then as basis for the initial microbial control program. The program covers the facility or product's life cycle and may be adapted following trend results and changes made in, for example, the facility design or process. It is recommended that on a regular basis (e.g. every two to three years or after relevant changes), the microbial control program is evaluated and if deemed necessary adapted. Trending of monitoring data strongly supports such evaluation (refer to Chapter 10 for a detailed review on trending of microbiological data). The following sections summarize the different controls to be included in a comprehensive microbial control program for non‐sterile products.
1.3 Main Factors to Be Controlled
1.3.1 Controlled Facilities
Current good manufacturing practices require that facilities manufacturing medicinal products for human use should be effectively designed:
- to run operations in a controlled environment
- to prevent product cross‐contamination
- to prevent microbiological contamination
- to provide sufficient space allowing operations running as intended
- to facilitate product, personnel, and material flow
- to permit effective cleaning and disinfection.
Facility design is part of all regulatory guidelines on current Good Manufacturing Practice (cGMP). For instance, the EU EudraLex volume 4 chapter 5 states that for medicinal products manufacturing areas the technical measures may include:
- Dedicated manufacturing facility (premises and equipment);
- Self‐contained production areas having separate processing equipment and separate heating, ventilation and air‐conditioning (HVAC) systems. It may also be desirable to isolate certain utilities from those used in other areas;
- Design of manufacturing process, premises and equipment to minimize opportunities for cross‐contamination during processing, maintenance and cleaning;
- Use of “closed systems” for processing and material/product transfer between equipment;
- Use of physical barrier systems, including isolators, as containment measures;
- Controlled removal of dust close to source of the contaminant, e.g. through localized extraction;
- Appropriate use of air‐locks and pressure cascade to confine potential air.borne contaminant within a specified area;
- Minimizing the risk of contamination caused by recirculation or re‐entry of untreated or insufficiently treated air;
- Use of automatic clean in place systems of validated effectiveness;
- For common general wash areas, separation of equipment washing, drying and storage areas.
For the United States, the guidelines on facilities can be found in the U.S. Code of Federal Regulations such as 211.42 Design and construction and 211.46 Ventilation, air filtration, air heating, and cooling. Other guidance on facility design can be found in PIC/S, WHO (e.g. WHO 2011) or engineering technical documents such as, for instance, the ISPE Baseline guideline on solid oral dosage forms (ISPE 2016).
For production areas of non‐sterile products most regulatory guidelines do not enforce that the air cleanliness is classified in terms of particle concentration (e.g. as per ISO 14644‐1). Also, the cleanroom classification of the EudraLex Annex 1 does not apply. However, there are a number of country‐specific guiding documents that provide some requirements for microbiological air quality in non‐sterile manufacturing facilities:
- The Chinese FDA GMP guideline (CFDA 2010) requires that The exposed processing areas for oral liquid and solid preparations, drugs applied through tract (including recta), epidermal products, and other non‐sterile products, as well as the exposed processing areas for handling immediate packaging materials should be designed as Grade D according to requirements in Annex 1 of Good Manufacturing Practice (GMP) for sterile products. This would imply that the cleanrooms have to be classified with grade D maximum permitted number of particles equal to or greater than 0.5 μm of 3,520,000.
- A similar requirement is given in the Mexicana NOM‐059‐SSA1‐2013, where an ISO Class 8 is required for prep...