![]()
Chapter 1
Introduction to Next Generation Sequencing Technologies
Lloyd Lowa and Martti T. Tammib
aPerdana University Centre for Bioinformatics (PU-CBi),
Block B and D1, MAEPS Building, MARDI Complex,
Jalan MAEPS Perdana, 43400 Serdang, Selangor, Malaysia.
bBiotechnology & Breeding Department,
Sime Darby Plantation R&D Centre, Selangor, 43400, Malaysia.
A Brief History of DNA Sequencing
In 1962 James Watson, Francis Crick and Maurice Wilkins jointly received the Nobel Prize in Physiology/Medicine for their discoveries of the structure of deoxyribonucleic acid (DNA) and its significance for information transfer in living material.1 The secret of DNA in orchestrating living activities lies in the arrangement of the four bases (i.e. adenine, thymine, guanine and cytosine). The linear sequence of the four bases can be considered as the language of life with each word specified by a codon that is made up of three bases. It was an interesting puzzle to figure out how codons specify amino acids. In 1968, Robert W. Holley, HarGobind Khorana and Marshall W. Nirenberg were awarded the Nobel Prize in Physiology/Medicine for solving the genetic code puzzle. Now it is known that collection of codons direct what, where, when and how much proteins should be made. Since the discovery of the structure of DNA and the genetic code, deciphering the meaning of DNA sequences has been an ongoing quest by many scientists to understand the intricacies of life.
The ability to read a DNA sequence is a prerequisite to decipher its meaning. Not surprisingly then, there has been intense competition to develop better tools to sequence DNA. In the 1970s, the first revolution in DNA sequencing technology began and there were two major competitors in this area. One was the commonly known Sanger sequencing method2,3 and another was the Maxam–Gilbert sequencing method.4 Over time, the popularity of the Sanger sequencing method and its modifications grew so much that it overshadowed other methods until perhaps 2005 when Next Generation Sequencing (NGS) began to take off.
In 1977, Sanger and colleagues successfully used their sequencing method to sequence the first DNA-based genome, a ϕX174 bacteriophage, which is approximately 5375 bp.5 This discovery heralded the start of the genomics era. Initially, the Sanger sequencing method in 1975 used a two-phase DNA synthesis reaction.2 In the first phase, a DNA polymerase was used to partially extend a primer bound onto a single stranded DNA template to generate DNA fragments of random lengths. In phase two, the partially extended templates from the earlier reaction were split into four parallel DNA synthesis reactions where each reaction only had three of the four deoxyribonucleotide triphosphates (dNTPs; which is made up of dATP, dCTP, dGTP, dTTP). Due to a missing deoxyribo-nucleotide triphosphate (e.g. dATP), the DNA synthesis reaction would stop at its 3′ end position just one position prior to where the missing base was supposed to be incorporated. All of these synthetized DNA fragments could then be separated by size using electrophoresis on an acrylamide gel. The DNA sequence could be read off a radioautograph since its DNA synthesis happened with the incorporation of radiolabeled nucleotides (e.g. S-dATP).35
There were many problems with the initial version of the Sanger sequencing method that required further innovations before its widespread use and this scenario is akin to what is happening in the recent NGS technological developments. Some problems of the early Sanger sequencing method included the cumbersome two-phase procedures, only short length of a DNA sequence could be determined, the requirement of a primer meant some sequences of the template had to be known, hazardous radio labeled nucleotides were used and there was also no automated way to read off a DNA sequence. Sanger and colleagues rapidly improved on the method described in 1975 by eliminating the two-phase procedure with the use of dideoxynucleotides as chain terminators.3 Briefly, the improved method started with four reaction mixtures that already had the single stranded DNA template hybridized to a primer. In each reaction, the DNA synthesis proceeded with four deoxyribonucleotide triphosphates (one with radiolabeled nucleotide) and one dideoxynucleotide (ddNTP). Whenever a dideoxyribonucleotide was incorporated, the reaction terminated and thereby produced a mixture of truncated fragments of varying lengths. These DNA fragments were then separated by electrophoresis and then read off from aradioautograph. By adjusting the concentration of ddNTPs, chain termination can be manipulated to produce a longer sequence read.
To solve the requirement of knowing some template sequences for primer design, cloning was introduced. For example, the M13 sequencing vector is commonly used as a holder for DNA insert and known primers that bind to the vector sequence are available to be used to sequence the unknown DNA insert. One major innovation to the Sanger sequencing method is the replacement of radioactive labels with fluorescent dyes.6 Four different dye colour labels are available for the four dideoxynucleotide chain terminators and thus, DNA fragments that terminate at all four bases can be generated in a single reaction and thus analyzed on a single lane of acrylamide gel. The electrophoresis is coupled to a fluorescent detector that is also connected to a computer and thus sequence data can be automatically collected. In 1986, Applied Biosystems commercialized the first automated DNA sequencer (i.e. Model 370A) that is based on the Sanger sequencing method. For an animation of the Sanger sequencing method, the reader should refer to the Welcome Trust Sanger Institute (http://www.wellcome.ac.uk/Education-resources/Education-and-learning/Resources/Animation/WTDV026689.htm).
Due to limitations of the chain terminator chemistry and resolution of the electrophoresis method, the Sanger sequencing method is only capable of sequencing a read of about 500 to 800 bases long. Most genes and other interesting DNA sequences are longer than that. Therefore, a method is required to first break up a longer DNA molecule into fragments, sequence the individual fragments and then piece them together to create a contiguous sequence (i.e. contig). In one approach known as the shotgun sequencing, the long DNA fragment is randomly sheared and then cloned for sequencing.7 A computer program is then used to assemble the sequences by finding overlaps. It is challenging to find sequence overlaps when thousands to millions of DNA fragments are generated. The problem requires alignment algorithms and some notable examples of early work in this area include the Needleman-Wunsch algorithm8 and Smith-Waterman algorithm.9 Details on the bioinformatics involved in NGS alignment tools and sequence assembly are given in Chapters 4 and 6, respectively.
Next Generation Sequencing Technologies
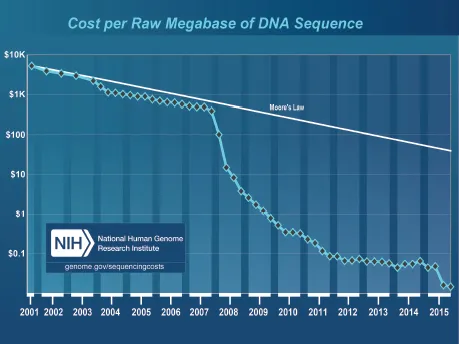
One of the goals of the Human Genome Project (HGP) is to support advancements in DNA sequencing technology.10 Although the HGP was completed with the Sanger sequencing method, many groups of researchers were already tinkering with new ideas to increase throughput and decrease cost of sequencing prior to the announcement of the first human genome draft in 2001. For example, developments for nanopore sequencing can be traced back to 1996 when researchers experimented with α-hemolysin.11 After years of experimentations, the second DNA sequencing technology revolution finally took off in 2005 and ended Sanger sequencing dominance in the marketplace. The revolution is still ongoing at the time of this writing and it can be seen from the rapid decline in the cost of sequencing since the introduction of NGS technologies (Figure 1).
The sequencing technologies associated with the second revolution are referred to by various names, including second generation sequencing, NGS and high throughput sequencing. It should perhaps be most appropriately termed as high throughput sequencing but NGS seems to be more commonly used to categorize such technologies and hence, this term is used for the book. For the purpose of this book, NGS technology refers to platforms that are able to sequence massive amount of DNA in parallel with a simultaneous sequence detection method and overall achieve a much cheaper cost per base than Sanger. These platforms include 454, ABI SOliD, Illumina and Ion Torrent. Due to the popularity of the Illumina platform at the time of this writing, the practical chapters (i.e. Chapters 3–10) of the book emphasize on the use of Illumina data as sample datasets.
Figure 1. The cost to sequence one million bases of a specified quality (i.e. a minimum Phred score of Q20 for Sanger sequencing and an equivalent of Q20 or higher accuracy for NGS data) according to the National Human Genome Research Institute (NHGRI).12 The cost of sequencing only made its rapid reduction in price from 2008 onwards.
There is a third revolution in sequencing technology underway with the commercialization of third generation sequencing technologies such as those from Pacific Biosciences and Oxford Nanopore Technologies. Third generation sequencing is defined as the sequencing of single DNA molecules without the need to halt between read steps, whether enzymatic or otherwise.13 There are three categories of single molecule sequencing: (i) sequencing by synthesis method whereby base detection occur real time (e.g. PacBio), (ii) nanopore technologies whereby DNA thread through a nanopore and are detected as they pass through it (e.g. Oxford Nanopore), and (iii) direct imaging of DNA molecules using advanced microscopy (e.g. Halcyon Molecular).
DNA sequence data generation process among different sequencing platforms may share similarities such as the general ‘wash and scan’ approach but they may differ in terms of cost, runtime and detection methods. The sequence data from different platforms have different characteristics such as error patterns and different tools being used to process the raw data to FASTQ format. Much of the internal workings of NGS sequencers are proprietary matters and users generally rely on providers to come out with their own tools for base calls as well as error calls. After that, a sequence is assumed as ‘correct’ and researchers proceed to analyze it. The subsequent sections aim to introduce the background and some details of commercially available platforms, which include 454, ABI SoliD, Illumina, Ion Torrent, PacBio, and Oxford Nanopore. Besides these six platforms, there are other companies out there that also innovate in this space such as SeqLL, GnuBIO, Complete Genomics and others, but they will not be covered here. For a list of available sequencing companies, readers are encouraged to read a news article by Michael Eisenstein in 2012 that was published by Nature Biotechnology, which detailed 14 NGS companies.14
454
A company named 454 Life Sciences Corporation made the first move in the NGS revolution. The company was initially majority owned by CuraGen. It was from this company that the name ‘454’ originated, which was just a code name for a project. 454 was later acquired by Roche in 2007. It made a public announcement in 2003 that it managed to sequence the entire genome of a virus in a single day.15 Then in 2005, scientists using 454 technology published an article in Nature on the complete sequencing and de novo assembly of Mycoplasma genitalium genome with 96% coverage and 99.96% accuracy in one run of the machine.16 In the same year, the company made a system named Genome Sequencer 20 (GS20) commercially available. This breakthrough in sequencing throughput and speed was an incredible feat when compared to the Sanger technology and it cre...