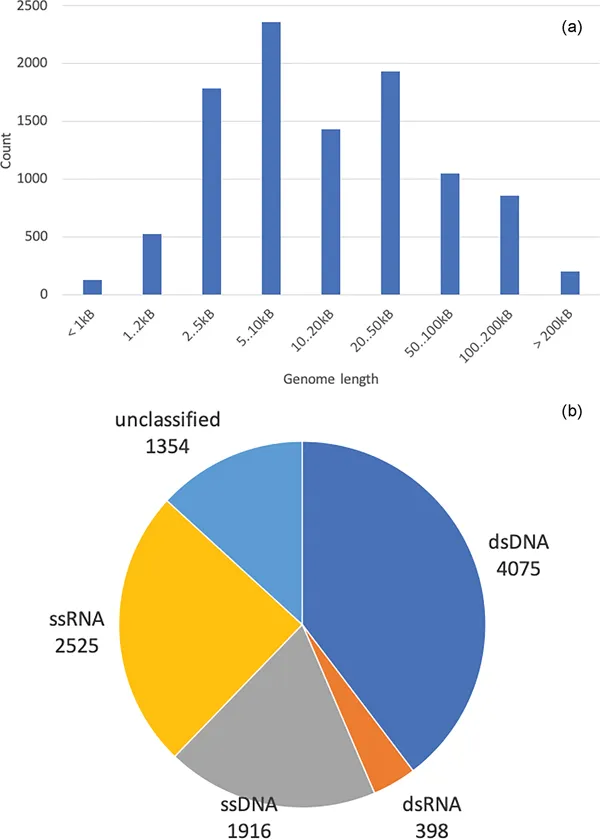
Viruses are the most numerous and deadliest biological entities on the planet, infecting all types of living organisms—from bacteria to human beings. The constantly expanding repertoire of experimental approaches available to study viruses includes both low-throughput techniques, such as imaging and 3D structure determination, and modern OMICS technologies, such as genome sequencing, ribosomal profiling, and RNA structure probing. Bioinformatics of viruses faces significant challenges due to their seemingly unlimited diversity, unusual lifestyle, great variety of replication strategies, compact genome organization, and rapid rate of evolution. At the same time, it also has the potential to deliver decisive clues for developing vaccines and medications against dangerous viral outbreaks, such as the recent coronavirus pandemics. Virus Bioinformatics reviews state-of-the-art bioinformatics algorithms and recent advances in data analysis in virology.
FEATURES
- Contributions from leading international experts in the field
- Discusses open questions and urgent needs
- Covers a broad spectrum of topics, including evolution, structure, and function of viruses, including coronaviruses
The book will be of great interest to computational biologists wishing to venture into the rapidly advancing field of virus bioinformatics as well as to virologists interested in acquiring basic bioinformatics skills to support their wet lab work.