Dmitrij Frishman, Manja Marz, Dmitrij Frishman, Manja Marz
This is a test
This is a test
Partager le livre
320 pages
English
ePUB (adapté aux mobiles)
Disponible sur iOS et Android
eBook - ePub
Virus Bioinformatics
Dmitrij Frishman, Manja Marz, Dmitrij Frishman, Manja Marz
Détails du livre
Aperçu du livre
Table des matières
Citations
À propos de ce livre
Viruses are the most numerous and deadliest biological entities on the planet, infecting all types of living organisms—from bacteria to human beings. The constantly expanding repertoire of experimental approaches available to study viruses includes both low-throughput techniques, such as imaging and 3D structure determination, and modern OMICS technologies, such as genome sequencing, ribosomal profiling, and RNA structure probing. Bioinformatics of viruses faces significant challenges due to their seemingly unlimited diversity, unusual lifestyle, great variety of replication strategies, compact genome organization, and rapid rate of evolution. At the same time, it also has the potential to deliver decisive clues for developing vaccines and medications against dangerous viral outbreaks, such as the recent coronavirus pandemics. Virus Bioinformatics reviews state-of-the-art bioinformatics algorithms and recent advances in data analysis in virology.
FEATURES
Contributions from leading international experts in the field
Discusses open questions and urgent needs
Covers a broad spectrum of topics, including evolution, structure, and function of viruses, including coronaviruses
The book will be of great interest to computational biologists wishing to venture into the rapidly advancing field of virus bioinformatics as well as to virologists interested in acquiring basic bioinformatics skills to support their wet lab work.
Foire aux questions
Comment puis-je résilier mon abonnement ?
Il vous suffit de vous rendre dans la section compte dans paramètres et de cliquer sur « Résilier l’abonnement ». C’est aussi simple que cela ! Une fois que vous aurez résilié votre abonnement, il restera actif pour le reste de la période pour laquelle vous avez payé. Découvrez-en plus ici.
Puis-je / comment puis-je télécharger des livres ?
Pour le moment, tous nos livres en format ePub adaptés aux mobiles peuvent être téléchargés via l’application. La plupart de nos PDF sont également disponibles en téléchargement et les autres seront téléchargeables très prochainement. Découvrez-en plus ici.
Quelle est la différence entre les formules tarifaires ?
Les deux abonnements vous donnent un accès complet à la bibliothèque et à toutes les fonctionnalités de Perlego. Les seules différences sont les tarifs ainsi que la période d’abonnement : avec l’abonnement annuel, vous économiserez environ 30 % par rapport à 12 mois d’abonnement mensuel.
Qu’est-ce que Perlego ?
Nous sommes un service d’abonnement à des ouvrages universitaires en ligne, où vous pouvez accéder à toute une bibliothèque pour un prix inférieur à celui d’un seul livre par mois. Avec plus d’un million de livres sur plus de 1 000 sujets, nous avons ce qu’il vous faut ! Découvrez-en plus ici.
Prenez-vous en charge la synthèse vocale ?
Recherchez le symbole Écouter sur votre prochain livre pour voir si vous pouvez l’écouter. L’outil Écouter lit le texte à haute voix pour vous, en surlignant le passage qui est en cours de lecture. Vous pouvez le mettre sur pause, l’accélérer ou le ralentir. Découvrez-en plus ici.
Est-ce que Virus Bioinformatics est un PDF/ePUB en ligne ?
Oui, vous pouvez accéder à Virus Bioinformatics par Dmitrij Frishman, Manja Marz, Dmitrij Frishman, Manja Marz en format PDF et/ou ePUB ainsi qu’à d’autres livres populaires dans Sciences biologiques et Microbiologie et biologie moléculaire. Nous disposons de plus d’un million d’ouvrages à découvrir dans notre catalogue.
1.1.6 Database Resources for Virus Genome Sequences
1.2 Comparison of Virus Genome Sequences
1.3 Protein Families and Orthologous Groups of Viruses
1.4 Evolution of Protein Families within Virus and Host Genomes
1.5 Outlook
References
1.1 Genomics of Viruses
1.1.1 Genome Types, Sizes, and Nomenclature
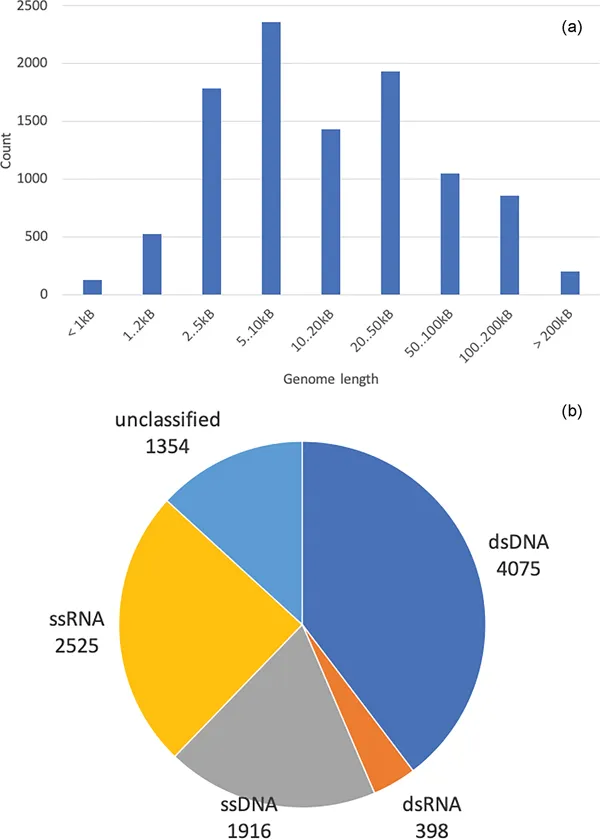
All viruses carry genetic information, which is encoded in genomic sequences. As a consequence of their host-based replication cycles, viruses usually have small or even very small genomes. However, the largest virus genome sequence can comprise more than a million nucleotides, which already is a typical size for small prokaryotic genomes (Figure 1.1a). Most eukaryotic genomes are based on single-stranded RNA (ssRNA), whereas most viruses of Bacteria and Archaea consist of double-stranded DNA (dsDNA) (Figure 1.1b). Despite their molecular structures, virus genome sequences are archived, exchanged, and computationally analyzed in the same one-letter IUPAC single-stranded nucleotide encoding as any cellular genome sequences. In the case of double-stranded genomes, canonical base pairing is assumed and one of the strands is selected for the genome sequence string. As both strands of a double-stranded genome are equivalent in terms of encoded information, the strand for the genome sequence of a newly sequenced virus genome is usually selected according to existing genome records in public sequence databases.
FIGURE 1.1 (a) Size distribution of virus genomes in NCBI RefSeq version 201 (O’Leary et al. 2016). (b) Genome types single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), single-stranded DNA (ssDNA), and double-stranded DNA (dsDNA) in NCBI RefSeq version 202 (O’Leary et al. 2016).
1.1.2 Genome Sequences from Cultures
Virus genomes can be sequenced from genetic material that is extracted from cultures of viruses in their host cells. These can be natural host cells as well as cell lines that are suitable for virus replication in the lab. The genomic material is extracted from the sample using standardized protocols. Ready-to-use kits for this purpose are commercially available for many viruses. However, their use for novel viruses often requires adaptation and validation. Virus genomic material is separated from host genomes and host transcripts by their different chemical makeup (e.g., single-stranded RNA vs. double-stranded DNA) and their different size and molecular weight, respectively.
The sequencing of virus genomes from cultures of many host cells rarely targets one uniform, static genome variant. Instead, a mixture of heterogeneous genome sequences is expected as a result of in-host evolution. This phenomenon is mostly remarkable in single-stranded RNA genomes, according to the limited error control during virus replication. Although the concept of “Quasispecies” initially described the effect of in-host evolution on fitness landscapes (Swetina and Schuster 1982), it is well-supported by recently collected genomic evidence from many viruses (Schuster 2016). Genome sequencing projects need to consider this phenomenon by their experimental design and their selection procedures for the genomic material. Specific genome assembly approaches allow the reconstruction of virus quasispecies genomes from deep short-read sequencing (Topfer et al. 2014). Recently developed sequencing techniques allow for long-read sequencing of complete virus genomes in single reads and provide a direct approach to the genome sequence diversity within a virus quasispecies (Yamashita et al. 2020).
Not always viral genomes are sequences on purpose. They can be sequenced along with host genomes and transcriptomes and are then usually removed and discarded. Furthermore, the presence of viruses in lab cell cultures and reagents is due to the abundance and diversity of viruses (Thannesberger et al. 2017).
1.1.3 Genomes from Environmental Samples
Virus genome sequences can also be obtained without cultivation, which is referred to as “metagenomics.” There are many reasons for using this approach, such as the survey for unknown viruses, the assessment of natural in-host evolution, the attempt to quantify natural abundances, or simply the lack of a suitable cultivation method. Metagenomic sequencing is performed on the material extracted from an environmental sample, which includes isolates from single multicellular individuals. It can be applied to RNA and DNA viruses and results in a mixture of virus and cellular reads, depending on the extraction and separation protocols, sequencing technique, and sequencing depth (Greninger 2018, Schulz et al. 2020).
The computational analysis of viral metagenomes from short reads is usually performed in specific workflows. These first assemble the reads into contigs or scaffolds using assembly software that is aware of different abundance of reads from different species. Assemblies from different assemblers or from different samples can be merged into one single metagenome assembly (Olm et al. 2017). Scaffolds are grouped into metagenomic bins by their relative sequence read depth in different samples or different genome extractions and by the similarity of their oligonucleotide frequency profiles. Compared to the binning of cellular metagenomes, no universally conserved, single-copy marker genes can be used for the binning of virus metagenomic assemblies. Consequently, also no general approach for the assessment of completeness, heterogeneity and contamination of virus metagenomic bins could be developed so far. Minimum Information about any (x) Sequence (MIxS) standard has recently been developed for reporting sequences of uncultivated virus genomes (Roux et al. 2019).
1.1.4 Proviruses
A special group of viruses is proviruses, which are integrated into their host’s genomes. Proviruses can be essential for the replication of viruses or can comprise latent forms of viruses. Both cases are relevant for virus genomics. Endogenous retroviruses make up significant portions of eukaryotic genomes, and prophages are frequently found in bacterial genomes. Proviruses are annotated according to their sequence characteristics in their host genomes, which can be combined with the prediction of whether the proviruses are still functional or degenerate. In genome assembly, the classification of viral contigs as provirus or viral contamination is challenging and requires the resolution of genomic repeats. Specific nomenclature for annotated retroviruses has been recently suggested (Gifford et al. 2018). Proviruses in microbial sequences can be automatically annotated based on their insertion sequence characteristics and their typical genome contents and gene order (Roux et al. 2015).
1.1.5 Annotation of Virus Genomes
The diversity of viral species, their life cycles, their genome structures, and their cultivability remain massive challenges for the development of universal software solutions for the annotation of virus genomes. Therefore, the main principles of automatic annotation of virus genomes are the detection of coding sequences by their oligonucleotide (such as codon) frequencies as well as the homology-based transfer of features and functional classifications from annotated genomes to newly sequenced genomes (Shean et al. 2019).
1.1.6 Database Resources for Virus Genome Sequences
The International Nucleotide Sequence Database Collaboration (INSDC) organizes the database resources that store newly sequenced and published genome sequences. It is a joint initiative of DDBJ, EMBL-EBI, and NCBI. INSDC has defined database and record structures for annotated genomes as well as partial genomic sequences, raw assemblies, and unassembled sequence reads. It includes formats for the attachment of functional annotation as well as contextual information relating to samples and experimental configurations (Cochrane et al. 2016).
Further databases with particular importance for virus genomes are NCBI RefSeq (O’Leary et al. 2016) and Uniprot/Swissprot (UniProt 2019). Whereas RefSeq is specialized in the selection and representation of complete genome sequences, SwissProt makes massive efforts in the manual curation of the annotations of viral gene products. Along with these efforts, ViralZone has been developed as a user-friendly knowledge base about virus genomics, including their virion structure, replication cycle, and host-virus interactions (Masson et al. 2013).
1.2 Comparison of Virus Genome Sequences
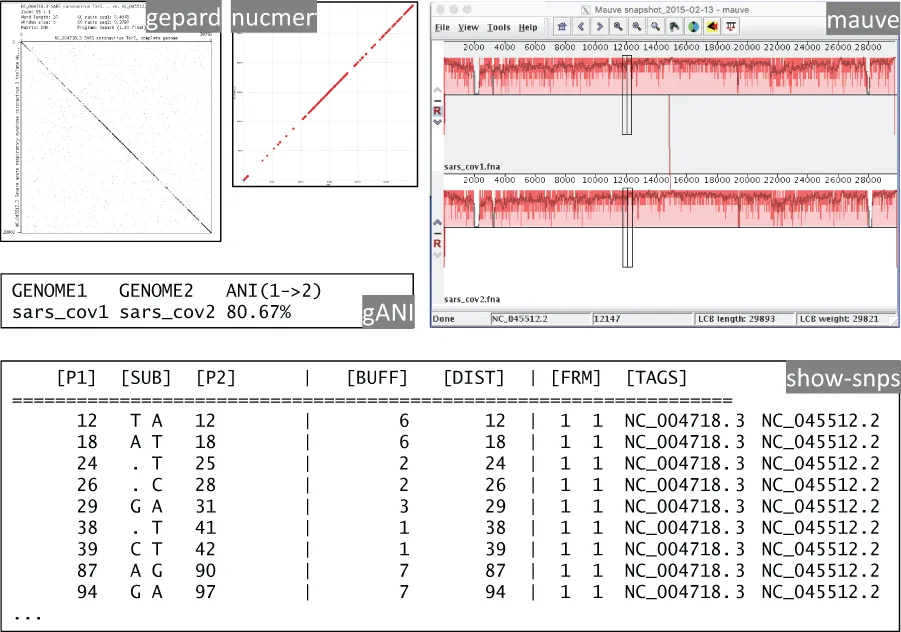
The most direct approach to comparative genomics of viruses is the direct comparison of genome sequences to each other. This can be performed for whole genomes, genome fragments, short subsequences, and single nucleotides (Figure 1.2).
FIGURE 1.2 Demonstration of the comparison of the same two genomes with Mauve, nucmer (Mummer), gepard, gANI, and show-snps (Mummer).
For comparisons of whole genomes, alignment-based methods and alignment-free methods exist. Alignment-based tools typically calculate regions of local similarity and subsequently extend these into whole genome alignments and visualizations (Darling et al. 2004). Alignments of partial or complete genomes can be calculated using generic methods, which utilize short common subsequences (Marcais et al. 2018). In order to account for the dynamics of genome evolution, which quickly leads to genome sequence divergence, additional constraints can be exploited to improve the accuracy of genome alignments, such as the sequence of codons and their encoded amino acids (Libin et al. 2019). For the comparison of many genomes, the similarities can be expressed numerically, e.g., as average nucleotide identity values (Varghese et al. 2015).
Alignment-free methods for genome alignment can focus on the visualization of genome similarities, which is particularly helpful to intuitively understand phenomena of genome evolution, such as transversions, inversions, and duplications (Krumsiek, Arnold, and Rattei...