Biophysical techniques are used in many key stages of the drug discovery process including in screening for new receptor ligands, in characterising drug mechanisms, and in validating data from biochemical and cellular assays.
This book provides an overview of the biophysical methods applied in drug discovery today, including traditional techniques and newer developments. Perspectives from academia and industry across a spectrum of techniques are brought together in a single volume. Small and biotherapeutic approaches are covered and strengths and limitations of each technique are presented. Case studies illustrate the application of each technique in real applied examples. Finally, the book covers recent developments in areas such as electron microscopy with discussions of their possible impact on future drug discovery.
This is a go-to volume for biophysicists, analytical chemists and medicinal chemists providing a broad overview of techniques of contemporary interest in drug discovery.

- 320 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Biophysical Techniques in Drug Discovery
About this book
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
CHAPTER 1
Impact and Evolution of Biophysics in Medicinal Chemistry
a Discovery Chemistry Research & Technology, Lead Generation and External Innovation, Eli Lilly and Company, Lilly Corporate Center, Indianapolis, IN 46285, USA
b Discovery Chemistry Research & Technology, Quantitative Biology, Eli Lilly and Company, Lilly Corporate Center, Indianapolis, IN 46285, USA
1.1 Introduction
Fifty five years ago, Max Perutz and John Kendrew received the 1962 Nobel Prize in Chemistry. Their pioneering work delivered the first 6 Å structure of the protein Myoglobin, and represented the foundation for modern structural biology.1–3 This first atomic-level picture heralded the complexity of the protein universe and provoked questions about how to predict the driving forces that allow proteins to rapidly fold into biologically relevant conformations. This was the beginning of biophysics. In 1969, the first enzyme structure of the extracellular nuclease of Staphylococcus Aureus as an enzyme-inhibitor complex was solved by F. Albert Cotton and coworkers.4
Thus began significant motivation to devise and evolve computational methods to predict protein folding. “Force fields” in modern computer simulation draw their information primarily from the ∼40 000 protein families represented in the Protein Data Bank (PDB), where structures obtained by X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy and, increasingly, cryo-electron microscopy (cryo-EM) are deposited. Today, X-ray crystallography has been extended further to characterise inherently disordered proteins and protein aggregation.5–8
Analysis of structural information has been applied to study protein equilibria and dynamics. The driving forces of (1) hydrogen bonding, (2) van der Waals interactions, (3) backbone conformational preferences, (4) electrostatics, and (5) hydrophobic interactions leading to the precise folding of proteins produce the minimum energy conformation, as can be seen crystallographically.9 Beyond the minimum energy conformation determined through x-ray crystallography, additional lower energy minima representing the dynamic movement of proteins can be studied by other biophysical techniques such as protein NMR and proton deuterium exchange mass spectrometry (HDX-MS).
Small molecules must be identified and optimized to productively interact with the protein's disease-relevant conformation or surface. This requires several pieces of information: (1) disease biology, (2) an understanding of the protein form or forms within the cell, and (3) an understanding of the protein surface and the potential interactions and MOA that can occur with the small molecule. Many of the same forces that impact protein conformation influence the low energy conformations and physical properties of drugs and can be assessed through small molecule crystallography, analytical methods and computational minimizations. Ultimately, the small molecule design team, the chemist, computational scientist, and structural biologist are challenged to identify opportunities for optimal molecular recognition from the protein. Optimization requires an understanding of interaction geometries and approximate contributions identified through crystal structures and demonstrated through affinity data.10 However, molecular interactions behave in a highly “non-additive fashion” and are context-dependent. Solvation of the protein and small molecule, long-range interactions and conformational changes of the protein (see Figure 3, ref. 9) all influence binding energetics.
Design of the small molecule must focus on specific intermolecular reactions based on available structural information. The most important and well defined are hydrogen bond interactions directly with the protein or through structural water.11–13 Weaker hydrogen bonds are also sometimes available when an aromatic ring can act as H-bond acceptor.14 Although H-bonds are among the strongest, these interactions should not be the sole focus of the designer. Orthogonal multipolar interactions of C–F to C=O such as the Bürgi–Dunitz angle,15 halogen bonds that leverage sigma-hole anisotropy, and cation–Pi interactions are also opportunities in the design of a molecule.16
Ultimately, the amount of hydrophobic surface buried through Van der Waals interactions upon ligand binding appears to best correlate with binding affinity.17 This concept holds for a diverse set of protein–ligand complexes, including protein–protein interactions.18 All other types of interactions are highly context-dependant and their utility must be tested within a given target. The thermodynamics and kinetics of the target interactions can be evaluated to understand progress and will be discussed in a later section.
Intramolecular interactions within a protein as well as stabilizing intermolecular interactions with an inhibitor were key to delivering the first biophysical technique, X-ray crystallography. The power of interpreting a low-energy atomic-level picture of a molecule bound to an enzyme created a paradigm shift in drug discovery. However, the dynamic nature of proteins requires an understanding of both their minimum energy states and also the status of their movement in other conformations. Further, these states must be measured and interpreted amidst the complex and dynamic processes in the cell. Techniques such as molecular imaging can provide direct measurement of the location and progression of processes. Connecting these techniques to non-invasive measurements of patient disease pathology and response in the evaluation of neurodegeneration,19 cardiovascular disease20 and oncology21,22 can leverage molecular imaging through PET-CT and MRI scans.
Biophysics is utilized throughout drug discovery strategies from target identification and selection, construct selection, screening, ligand optimization, drug development and ultimately imaging within our patients. The following sections will provide context for the techniques presented in this book.
1.2 Evolution of Biophysics in Medicinal Chemistry
1.2.1 Phenotypic Drug Discovery
The earliest drug identification and approval were driven by small molecule phenotypic drug discovery (PDD) approaches.23–25 This strategy involved identifying and optimizing efficacy in animal physiological and behavioural models for human diseases, and ex vivo tissue-based or phenotypic cellular assays. While many drugs used clinically today were discovered agnostic of their target protein, work in this fashion increased the necessary investment in the systematic iteration and synthesis of lead molecules focusing on potency and phenotype. Without specific target and mechanism of action information, agents often displayed poly-pharmacology, which could be good for disease modification or bad for toxicology. Coupled with extended times in early stage discovery, the advent of protein crystallography, and the potential for off-target liabilities that wouldn't be identified until entry into the clinic, drug discovery attempted to minimize these challenges through targeted therapies, that is, target directed drug discovery (TDD) (Figure 1.1).26
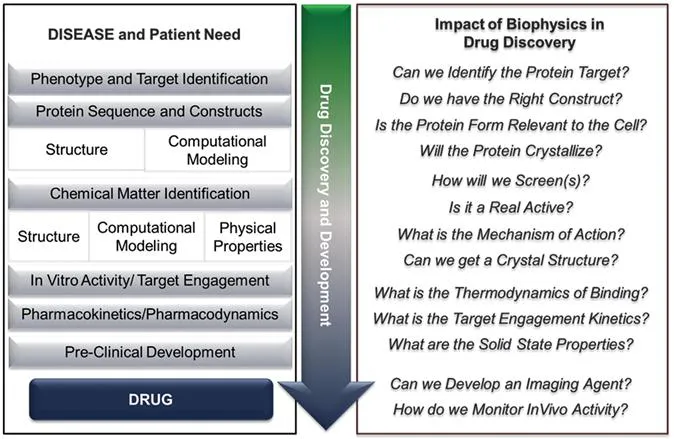
Figure 1.1 Drug discovery pathway to a medicine with examples of key questions that biophysics can address experimentally and influence in molecule identification, progression and design.
1.2.2 Targeted and Fragment-based Drug Discovery
With the advent of a deeper understanding of the proteins involved in a disease pathway through cell biology tools such as siRNA, proteomics, knock-outs, and human genetics (mutations found in patient populations), drug discovery efforts have focused more on specific proteins believed to be involved in a disease. This knowledge has led to a paradigm shift in drug discovery that went from dosing a limited set of compounds in animal models to screening 250 000 to 1 million compounds in biochemical assays in a specific protein. Typically, the biochemical assays were performed in formats that looked at competition binding or enzyme activity against recombinant proteins.
Drug discovery targets, in response, moved into more complex protein targets such has epigenetics, protein–protein interactions (PPI), membrane-bound proteins and transporters, and protein aggregation. This resulted in gaps or greater challenges in screening approaches, and protein expression, production and purification. The new target space ultimately affected structural biology approaches. As an example, discrete protein target EZH2 (Section 1.5.2), actually a component of a complex of proteins, requires this complex for activity and stability. In addition, protein complexes could in theory be inhibited by binding in different proteins and read-out as competitive, non-competitive, uncompetitive, mixed or irreversible in nature. In the case of protein–protein interactions, one may simply be interacting with a protein and disrupting a productive, functional interaction with a second protein.
This paradigm shift in targets also resulted in re-thinking how compound libraries were computationally selected for screening. For example, PPI targets required larger molecules that were not present in most traditional libraries based on Lipinki's rules,24 and when starting points were found in traditional screening, there was very little room to maintain potency while fixing the physical properties. To overcome gaps in compound collections and property challenges, some drug discovery groups implemented a new strategy and proposed to screen compound “fragments” with MW<300 Da. Fragment-based drug discovery (FBDD) takes advantage of Jencks’ description of Gibb's free energy and attributes of the binding energy between enzyme and substrate.25 By beginning with fragments, a greater chemical space could be sampled with smaller sets of molecules while optimizing biological activity and physical properties in parallel.26 This introduced a strategic shift in how to approach drug discovery and an opportunity to identify chemical matter without a large compound collection.
The challenge with FBDD is that the initial compound actives have highly efficient per heavy atom (non-hydrogen) target binding but low measured binding constants. Given their low affinity, high concentration biochemical assays (enzyme or competition binding) were used, but did not behave or were still not sensitive enough to pick up fragments in all targets or target classes. Sensitive biophysical methods had to be identified. Teams also needed orthogonal methods to confirm the fragment as a legitimate active, identify a potential MOA, and to provide clues about vectors in order to decorate the molecule for efficient, key interactions within the larger protein active site.
1.2.3 Phenotypic Drug Discovery 2.0
A renewed interest in PDD strategies was influenced by a focus on the complexity of patients and their disease states in oncology, autoimmune disease and neurodegeneration. Coupled with technological developments at the interfaces of disciplines scientists have been able to deliver complex cell culture systems and gene-edited disease relevant mutations for phenotypic screening assays.27–29 Advances in mass spectrometry methods such as affinity selection (AS-MS), cellular thermal shift assays (CETSA) and cDNA expression microarray technologies have allowed probing of complex pathways. Improvements in cell-based and model-organism based automated screens,30–32 along with the development of new biophysical methodologies have enabled improved screening, faster identification of targets and profiling of the MOA in complex biological samples at the genomic, proteomic and phenotypic levels.33
1.2.4 Evolving Compound Collections and Chemical Technologies
Highly elaborate compounds that became part of compound collections were de...
Table of contents
- Cover
- Title
- Copyright
- Contents
- Chapter 1 Impact and Evolution of Biophysics in Medicinal Chemistry
- Chapter 2 Ligand-detected NMR Methods in Drug Discovery
- Chapter 3 Receptor-based NMR Techniques in Drug Discovery
- Chapter 4 Molecular Mechanisms of Drug Action: X-ray Crystallography at the Basis of Structure-based and Ligand-based Drug Design
- Chapter 5 Mass Spectrometry in Biophysics: from High Throughput Screening to Structural Biology
- Chapter 6 Characterization of Pharmaceutical Solids Combining NMR, X-ray diffraction and Computer Modelling
- Chapter 7 Surface Plasmon Resonance for Identifying and Characterising Small Molecule Ligands
- Chapter 8 Fluorescent Thermal Shift Assays for Identifying Small Molecule Ligands
- Chapter 9 Fluorescent Probes in Medicinal Chemistry
- Chapter 10 Transmission Cryo-electron Microscopy in Drug Discovery
- Chapter 11 Molecular Imaging
- Subject Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Biophysical Techniques in Drug Discovery by Angeles Canales in PDF and/or ePUB format, as well as other popular books in Biological Sciences & Biochemistry. We have over 1.5 million books available in our catalogue for you to explore.