Jetzt in der dritten, uberarbeiteten, erweiterten, deutschsprachigen Auflage!
Dieses NMR-Lehrbuch bietet dem Leser eine praxisnahe Hilfe bei der Ubersetzung von NMR-Spektren in die Struktur organischer Verbindungen. Nach einer sehr kurzen theoretischen Einleitung, in der die wichtigsten Begriffe erklart werden, folgt eine Einfuhrung in die Strategie und Taktik der Strukturaufklarung mit NMR-Methoden. Ein Schwerpunkt sind 55 Fallstudien mit Spektren bzw. Spektrenserien abgestuften Schwierigkeitsgrades zum Selbststudium sowie ausfuhrliche Losungsvorschlage zum Nachvollziehen.
Neu hinzugekommen sind einige aktuelle zweidimensionale NMR-Experimente der heteronuklearen und homonuklearen Korrelation chemischer Verschiebungen sowie neue Aufgaben zu deren Anwendung. Dieser Klassiker unter den NMR-Lehrbuchern ist unentbehrlich fur jeden, der zur Aufklarung organischer Strukturen die NMR-Spektroskopie anwendet.

- German
- ePUB (handyfreundlich)
- Über iOS und Android verfügbar
eBook - ePub
Vom NMR-Spektrum zur Strukturformel organischer Verbindungen
Über dieses Buch
375,005 Studierende vertrauen auf uns
Zugang zu über 1 Million Titeln zu einem fairen monatlichen Preis.
Mit unseren Lerntools kannst du noch effizienter lernen.
Information
1
Grundbegriffe, Meßgrößen, Meßverfahren in Kürze
1.1 Chemische Verschiebung
Als chemische Verschiebung bezeichnet man die Abängigkeit der Larmorfrequenz eines Kernspins von seiner chemischen Umgebung. Der Kernspin ist der mechanische Drehimpuls des Atomkerns, die Larmorfrequenz seine Präzessionsfrequenz im statischen Magnetfeld (Abb. 1.1). Sie ist der Kraftflußdichte B0 des Magnetfelds proportional (vo/B0 = const.) 1-3. Praktischerweise bezieht man die chemische Verschiebung z.B. der Protonen auf den Standard Tetramethylsilan [TMS, (CH3 )4Si] und nicht auf das Proton H+, d.h. man mißt die Frequenzdifferenz (Hz) zum Signal des Standards (Tetramethylsilan,TMS, für lH- und 13C-NMR) und teilt diese durch den der Kraftflußdichte B0 des Magnetfelds proportionalen Absolutwert der Larmorfrequenz des Standards (z.B. 400 MHz für die Protonen und 100 MHz für die 13C-Kerne des TMS im Falle eines 400 MHz- Spektrometers mit B0 = 9.4 Tesla). Der Quotient definiert den vom Magnetfeld unabhängigen δ- Wert der chemischen Verschiebung. Dabei wird eine Frequenzdifferenz in Hz durch eine Frequenz in MHz geteilt; da sich beide Frequenzen wie 1:106 verhalten, wird δ oft in ppm (part per million) angegeben.
Abb. 1.1. Kernpräzession: Kernladung und Kernspin verleihen Atomkernen wie Protonen und Kohlenstoff-13 ein magnetisches Moment. Der Vektor μ des magnetischen Moments präzessiert im statischen Magnetfeld mit der Larmor-Frequenz v 0 um die Richtung des magnetischen Kraftflußdichte-Vektors B0
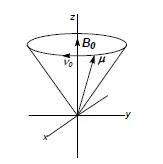
Ursache der chemischen Verschiebung ist u.a. die Abschirmung der Kernspins im Molekül durch die Elektronenhülle. Die Elektronen erzeugen ein Abschirmfeld, das dem äußeren Magnetfeld entgegengerichtet ist, daher die Präzessionsfrequenz der Kernspins, also ihre chemische Verschiebung verkleinert. Man bezeichnet einen Atomkern (z.B. ein Proton) mit kleiner Verschiebung als abgeschirmt (hohes Abschirmfeld); ein Kern mit großer Verschiebung ist dagegen entschirmt (kleines oder tiefes Abschirmfeld, Abb. 1.2).
1.2 Spin-Spin-Kopplung und Kopplungskonstanten
Die indirekte oder skalare Spin-Spin-Kopplung von Kernspins über kovalente Bindungen verursacht die Aufspaltung von NMR-Signalen zu Multipletts in der hochauflösenden NMR-Spektros-kopie gelöster Verbindungen. Die direkte oder dipolare Kopplung zwischen Kernspins durch den Raum wird nur in der Festkörper-Kernresonanz beobachtet. In Lösung mittelt die Molekülbewegung diese Kopplung aus.
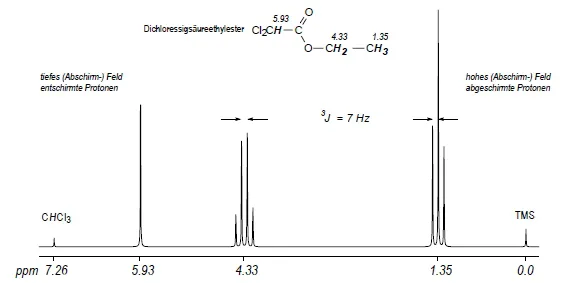
Die Kopplungskonstante ist in Spektren erster Ordnung (Abschn. 1.4) der Frequenzabstand J in Hz von zwei Multiplettübergängen. Im Gegensatz zum Frequenzbetrag der chemischen Verschiebung hängt die Kopplungskonstante nicht von der Magnetfeldstärke ab (Beispiel: Abb. 2.7). In der hochauflösenden NMR unterscheidet man zwischen Kopplungen über eine Bindung (1J oder einfach J, unmittelbare Kopplungen) und mehrere Bindungen, z.B. über zwei (2J, geminale Kopplungen), drei (3J, vicinale Kopplungen), vier und fünf Bindungen (4J und 5J, Fernkopplungen). Die CH2- und CH3-Protonen der Ethyl-Gruppe des Dichloressigsäureethylesters in Abb. 1.2 sind z.B. durch drei Bindungen getrennt; ihre (vicinale) Kopplungskonstante beträgt 3J = 7 Hz.
Abb. 1.2. 1H-NMR Spektrum des Dichloressigsäureethylesters (CDCI3, 25 °C, 80 MHz). Das Proton der CHCl2 Gruppe ist weniger abgeschirmt (stärker entschirmt) als die Protonen der CH 2- und CH 3-Reste
1.3 Signalmultiplizität (Multipletts)
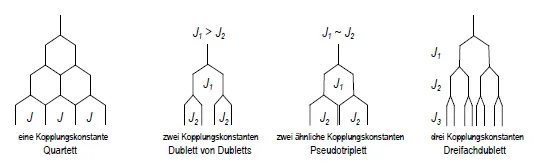
Die Signalmultiplizität in NMR-Spektren erster Ordung (Abschn. 1.4) ist der Aufspaltungsgrad eines NMR-Signals infolge der Spin-Spin-Kopplung. Signale ohne erkennbare Aufspaltung bezeichnet man als Singuletts (s), solche mit zwei-, drei-, vier-, fünf-, sechs-, siebenfacher Aufspaltung als Dubletts (d), Tripletts (t), Quartetts (q Abb. 1.2, 1.3), Quintetts (qui), Sextetts (sxt) und Septetts (sep), aber nur, wenn die einzelnen Signale des Multipletts gleichen Abstand besitzen, also nur eine Kopplungskonstante beteiligt ist. Verursachen zwei bzw. drei verschiedene Kopplungskonstanten ein Multiplett, so handelt es sich um ein Doppeldublett (dd) bzw. Dreifachdublett (ddd, Abb. 1.3). Sind die beiden Kopplungskonstanten eines Doppeldubletts zu ähnlich (J1 ~ J2), so überlappen die mittleren Signale; man beobachtet ein “Pseudotriplett” (“t”, Abb. 1.3).
Das 1H-NMR-Spektrum des Dichloressigsäureethylesters (Abb. 1.2) zeigt z.B. ein Triplett für die CH3-Gruppe (zwei vicinale H), ein Quartett für die OCH 2-Gruppe (drei vicinale H) und ein Singulett für das CHCl2-Fragment (kein vicinales H als Kopplungspartner).
Abb. 1.3. Quartett, Doppeldublett, Pseudotriplett und Dreifachdublett (Dublett von Dubletts von Dubletts)
1.4 Spektren erster und höherer Ordnung
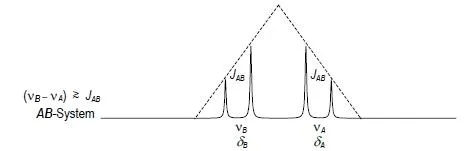
Multipletts erster Ordnung liegen vor, wenn die Kopplungskonstante klein im Vergleich zur Verschiebungsdifferenz der Kopplungspartner ist (vX–vA >> JAX). Man spricht dann von AmXm- Systemen, wobei Kern A die kleinere, Kern X die deutlich größere Verschiebung zugeordnet wird. Ein AX-System (Abb. 1.4) besteht aus dem A-Dublett und dem X-Dublett mit der gemeinsamen Kopplungskonstanten JAX.
Abb. 1.4. Zweispinsystem vom Typ AX mit großer Verschiebungsdifferenz im Vergleich zur Kopplungskonstanten (schematisch)
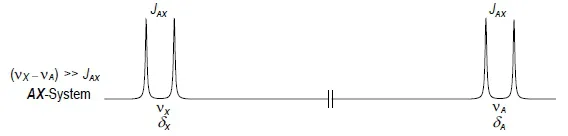
Abb. 1.5. Relative Intensitätsverhältnisse von Multipletts erster Ordnung (Pascalsches Zahlendreieck)

Multiplizitätsregeln gelten für Spektren erster Ordnung (AmXm-Systeme): nX koppelnde Kernspins X mit Kernspin-Quantenzahl I = 1/2 bewirken eine (nX +l)-fache Aufspaltung des A-Signals; die Intensitäten der einzelnen Signale eines Multipletts erster Ordnung folgen dem Pascalschen Zahlendreieck (Abb. 1.5). Die Protonen der Ethyl-Gruppe in Dichloressigsäureethylester (Abb. 1.2) bilden z.B. ein A3X2-System mit der Kopplungskonstanten 3JAX = 7 Hz; die A-Protonen (mit der kleineren Verschiebung) spalten in ein Triplett t auf (zwei vicinale Protonen X, nX +l = 3); die X- Protonen bilden wegen der drei vicinalen A-Protonen ein Quartett (nA + 1 = 4). Für nX koppelnde Kernspins mit beliebigen Kernspinquantenzahlen IX zeigt das A-Signal eine (2nXIX +l)-fache Aufspaltung (Beispiel: Abb. 1.9).
Multipletts (Spektren) höherer Ordnung liegen vor, wenn die Kopplungskonstante die Größenordnung der Verschiebungsdifferenz der Kopplungspartner erreicht. Man spricht dann von AmBn- Systemen, wobei Kern A die kleinere, Kern B die größere Verschiebung zugeordnet wird.
Ein AB-System (Abb. 1.6) besteht z.B. aus dem A-Dublett und dem B-Dublett mit der gemeinsamen Kopplungskonstanten JAB , wobei die äußeren Signale beider Dubletts geschwächt, die inneren verstärkt werden. Man spricht vom AB-Effekt, einem zum Zentrum des AB-Systems symmetrischen “Dacheffekt”. Dieser wird in Protonen-NMR-Spektren beobachtet (Abb. 1.2, Ethyl-Quartett und Triplett), wenn die Magnetfeldstärke der Bedingung (VX-vA >> JAX ) nicht hinreichend genügt. Die chemischen Verschiebungen vA und vB entsprechen dann nicht mehr den Zentren sondern den Schwerpunkten der beiden Dubletts; sie nähern sich den intensiveren inneren Signalen.
Abb. 1.6. Zweispinsystem vom Typ AB mit kleiner Verschiebungsdifferenz im Vergleich zur Kopplungskonstanten (schematisch)
1.5 Chemische und magnetische Äquivalenz
Chemische Äquivalenz: Atomkerne in gleicher chemischer Umgebung sind chemisch äquivalent, zeigen daher dieselbe chemische Verschiebung. Die 2,2’- und 3,3’-Protonen eines 1,4-disubstituierten Benzen-Rings sind z.B. aus Gründen der Molekülsymmetrie chemisch äquivalent.
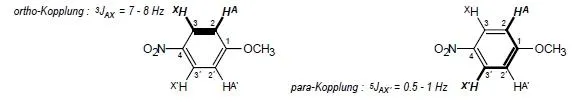
Magnetische Äquivalenz: Chemisch äquivalente Atomkerne sind magnetisch äquivalent, sofern sie mit allen anderen Kernspins des Moleküls dieselben Kopplungskonstanten aufweisen. Die 2,2’- (AA’-) und 3,3’- (X,X’-)-Protonen eines 1,4-disubstituierten Benzen-Rings wie 4-Nitroanisol sind z.B. magnetisch nicht äquivalent, weil das 2-Proton A mit dem 3-Proton X eine ortho- (etwa 7-8 Hz), mit dem 3’-Proton X’ dagegen eine para-Kopplung (etwa 0.5 bis 1 Hz) aufweist. Man spricht daher weder von einem A2X2- noch von einem (AX)2-, sondern von einem AA’ XX’-System (Beispiel: Abb. 2.6). Das 1 H-NMR-Spektrum ist in diesem Fall nicht erster Ordnung; ebensowenig gelten die Multiplizitätsregeln. Die Methyl-Protonen des Dichloressigsäureethylesters (Abb. 1.2) sind dagegen chemisch und magnetisch äquivalent, da die 3 JHH -Kopplungskonstanten von den geometris...
Inhaltsverzeichnis
- Cover
- Series page
- Title page
- Copyright
- Vorwort
- Symbole und Abkürzungen
- 1: Grundbegriffe, Meßgrößen, Meßverfahren in Kürze
- 2: Erkennung von Strukturmerkmalen durch NMR
- 3: Probleme
- 4: Problemlösungen
- Bibliographie
- Formelverzeichnis der Problemlösungen
- Sachverzeichnis
Häufig gestellte Fragen
Ja, du kannst dein Abo jederzeit über den Tab Abo in deinen Kontoeinstellungen auf der Perlego-Website kündigen. Dein Abo bleibt bis zum Ende deines aktuellen Abrechnungszeitraums aktiv. Erfahre, wie du dein Abo kündigen kannst
Nein, Bücher können nicht als externe Dateien, z. B. PDFs, zur Verwendung außerhalb von Perlego heruntergeladen werden. Du kannst jedoch Bücher in der Perlego-App herunterladen, um sie offline auf deinem Smartphone oder Tablet zu lesen. Erfahre, wie du Bücher herunterladen kannst, um sie offline zu lesen
Wir sind ein Online-Lehrbuch-Abo, bei dem du für weniger als den Preis eines einzelnen Buches pro Monat Zugang zu einer ganzen Online-Bibliothek erhältst. Mit über 1 Million Büchern zu über 990 verschiedenen Themen haben wir bestimmt alles, was du brauchst! Erfahre mehr über unsere Mission
Achte auf das Symbol zum Vorlesen bei deinem nächsten Buch, um zu sehen, ob du es dir auch anhören kannst. Bei diesem Tool wird dir Text laut vorgelesen, wobei der Text beim Vorlesen auch grafisch hervorgehoben wird. Du kannst das Vorlesen jederzeit anhalten, beschleunigen und verlangsamen. Erfahre mehr über die Funktion „Vorlesen“
Ja! Du kannst die Perlego-App sowohl auf iOS- als auch auf Android-Geräten nutzen, damit du jederzeit und überall lesen kannst – sogar offline. Perfekt für den Weg zur Arbeit oder wenn du unterwegs bist.
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Bitte beachte, dass wir Geräte, auf denen die Betriebssysteme iOS 13 und Android 7 oder noch ältere Versionen ausgeführt werden, nicht unterstützen können. Mehr über die Verwendung der App erfahren
Ja, du hast Zugang zu Vom NMR-Spektrum zur Strukturformel organischer Verbindungen von Eberhard Breitmaier im PDF- und/oder ePub-Format sowie zu anderen beliebten Büchern aus Scienze fisiche & Spettroscopia e analisi dello spettro. Aus unserem Katalog stehen dir über 1 Million Bücher zur Verfügung.