
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
About this book
Now in a revised edition, Comparative Pharmacokinetics: Principles, Techniques, and Applications presents the principles and techniques of comparative and veterinary pharmacokinetics in a detailed yet practical manner. Developed as a tool for ensuring that pharmacokinetics studies are properly designed and correctly interpreted, the book provides complete coverage of the conceptual basis of pharmacokinetics as used for quantifying biological processes from the perspectives of physiology and medicine. New chapters have been added on quantitative structure permeability relationships and bioequivalence, and a number of existing chapters have been significantly revised and expanded to provide a current resource for veterinary and comparative pharmacokinetics.
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
1
Introduction
Pharmacokinetics is best defined as the use of mathematical models to quantitate the time course of drug absorption and disposition in man and animals. With the tremendous advances in medicine and analytical chemistry, coupled with the almost universal availability of computers, what was once an arcane science has now entered the mainstream of most fields of human and veterinary medicine. This discipline has allowed dosages of drugs to be tailored to individuals or groups to optimize therapeutic effectiveness, minimize toxicity, and avoid violative tissue residues in the case of food-producing animals.
What differentiates this discipline from other fields of pharmacology and medicine is its focus on quantitating biological phenomena using various mathematical models and restricting its purview to the movement of drugs and chemicals into, through, and out of the body. The subsequent effects of these drugs on biological processes fall in the realm of pharmacodynamics (PD), which is beyond the scope of the present pharmacokinetic text but is extensively reviewed in Chapter 13 when linkage to pharmacokinetic models is developed. There are numerous applications of pharmacokinetics in clinical practice, some of them unknown to the practitioner as actually being pharmacokinetic modeling exercises since the terminology has become embedded into the lexicon of general medicine.
Since the publication of the first comparative pharmacokinetics text by Desmond Baggot in 1977, there has been explosive growth in all aspects of this discipline. This growth has continued after the publication of the first edition of the present text in 1999 and the release in 2004 of the pivotal UK “PK and PK-PD in Veterinary Medicine” workshop (Lees, 2004). The continued integration of pharmacokinetic concepts into global veterinary drug regulations further fuels this growth, a development that can be appreciated by reading Chapter 15 on regulatory aspects of drug product bioequivalence.
The primary pharmacokinetic models originally utilized by comparative and veterinary pharmacokineticists were the classic open compartmental models. These models, first clearly elucidated by Teorell in 1937, have been the mainstay of pharmacokinetics for much of the last decade. However, the use of noncompartmental models, especially those based on statistical moment analyses, has recently expanded across multiple areas. This popularity can be linked in part to a superb suitability for analysis by digital computers. Paradoxically, many of the properties that make the analysis of serum pharmacokinetic data amenable to exponential equations result from a few mathematical peculiarities in the solution of these compartmental models. Newer noncompartmental approaches to data analysis share many of these attributes and thus also share the same limitations of the classic modeling approaches. These intricacies will be completely explored in this text.
In the last decade, there has been a greatly increased use of so-called population pharmacokinetic approaches. This growth has been facilitated by the availability of user-friendly software and the implicit recognition that interindividual variability in the physiology underlying pharmacokinetic parameters may overshadow drug-specific parameters. Quantitating this variability using stochastic techniques is becoming widespread.
Pharmacokinetic principles have become widespread in the discipline of toxicology, an application termed toxicokinetics. There are no fundamental differences between pharmacokinetic and toxicokinetic principles except that the latter often deal with higher doses of chemicals, which may saturate metabolizing enzymes and in some cases may damage eliminating organs, thereby altering the disposition of the toxin. However, the principles involved are identical, and the concepts presented in this text are applicable to both fields.
Physiologically based pharmacokinetic (PBPK) models have become routine in many fields of pharmacology and toxicology. These models, unlike the others mentioned, build on the basis of sound anatomical and physiological principles and, although data-intensive, may allow the best opportunity for true mechanism-based interspecies pharmacokinetic extrapolations. Individual organ function is easily scaled across species and in vitro data may be extrapolated to the whole animal. This modeling approach has been increasingly applied to the problem of drug and chemical residues in food-producing animals.
The goal of quantitative pharmacology is always to extrapolate the drug concentration profile in the simpler in vitro experimental environment to that which actually exists in the cells or tissues of whole animals. Such an extrapolation (albeit very crude) is made daily with the use of minimum inhibitory concentrations (MICs) to estimate the efficacy of an antimicrobial drug against a specific bacteria in a human or animal patient. Recent work has focused on quantifying in vitro-to-in vivo correlations, one flavor of which termed IVIVC focuses on predicting oral absorption from in vitro dissolution studies. In vitro studies may be conducted with very simple subcellular, single-cell, or tissue culture systems or more complex perfused organ preparations (also referred to as ex vivo models).
In drug development and biochemical toxicology laboratories, extrapolation is often from a simple receptor or subcellular fraction assay, which detects drug or toxin binding, to the dose of drug that would be required to achieve this effective concentration in vivo. Alternatively, DNA binding or cytotoxicity screens may detect potential adverse events associated with a specific chemical. This defines a hazard in the risk assessment process. However, sufficient exposure in the intact organism is still required for this hazard to be realized as a risk. Pharmacokinetics is often the bridge in this extrapolation. In fact, sophisticated concentration–response relationships, obtained from in vitro bioassay systems, may be defined and then linked to the in vivo dose–response profile using integrated pharmacokinetic–pharmacodynamic (PK-PD) modeling techniques. PBPK models also provide the framework for tying drug delivery to cells in modern systems biology schemes that attempt to model the cellular responses seen after chemical exposure using the tools of genomics, proteomics, and metabonomics.
Work has also exploded in the field of quantitative structure–activity relationships (QSARs) that relates molecular properties to biological activity. From its application to pharmacokinetics, progress has been made to use such techniques to predict oral bioavailability or transdermal delivery. A new chapter in this edition introduces these concepts.
The extrapolation of pharmacokinetic parameters across species is a major focus of research. This is true in laboratory animal medicine and especially so in exotic animal and zoo animal medicine. Many “classic” compartmental pharmacokinetic studies conducted in multiple animal species have been extrapolated using the techniques of allometry. This is often employed when laboratory animal toxicology data must be extended to humans to put into perspective the relationship between the expected toxic dose and therapeutically useful doses. This later concept of a “therapeutic window” framed by a minimal effective therapeutic dose or resultant concentration and maximally safe toxic threshold is found in many areas of medicine and is implicitly based in pharmacokinetic methodology.
The fields of clinical pharmacology have grown in both human and veterinary medicine. Subpopulations of patients based on age or disease processes are routinely defined and dosages of drug appropriately altered. Part of the growth of this discipline was facilitated by the routine application of pharmacokinetics in clinical patients. This was facilitated by the development of population pharmacokinetic approaches mentioned earlier, which merge the estimation of pharmacokinetic parameters with simultaneous clinical estimates of physiological parameters and population variability. Its application to defining disease-induced changes in drug disposition and probing the nature of pharmacokinetic variance are widespread. In veterinary medicine, these same principles are needed to extrapolate dosage regimens for extralabel drug use.
This proliferation of pharmacokinetics throughout these diverse fields has been propelled by the explosive growth in analytical methodologies using principles of both chromatography (high-performance liquid and gas chromatography) and immunology (radioimmunoassay, enzyme-linked immunosorbent assay [ELISA]). Not only has the cost per sample of these procedures plummeted but their availability and sensitivity have also increased tremendously. For many drugs, simple disposable card-type assays are being developed that will provide the clinician instantly with estimates of drug concentrations. In veterinary medicine, such assays are available to monitor milk and urine for the presence of violative drug residues.
With the drug concentration data now readily available, complex mathematical modeling that was once restricted to the esoteric and truly “user-unfriendly” and even “user-adverse” mainframe computers can now be routinely done on nearly any available personal computer using one of a myriad of simple-to-use pharmacokinetic software packages including Win-Nonlin and other packages (e.g., CONSAAM and SAAM, PK-Analyst, P-Pharm). In fact, the proliferation of these automated software packages is one of the developments that highlighted the need for this text because, parallel to this proliferation of tools to conduct pharmacokinetic analyses, many workers have failed to study its basic principles and often inappropriately apply models to experimental and clinical situations.
1.1 OBJECTIVES AND PHILOSOPHY
The purpose of this book is to provide an introduction of the discipline of pharmacokinetics for the student, researcher, and comparative medicine clinician. The text presents an overview of the basic processes of drug absorption and disposition and then details how these processes can be quantitated using different pharmacokinetic approaches. The book is directed toward both the individual responsible for doing the analysis and the user of the pharmacokinetic information generated. To properly employ pharmacokinetic information, the limitations of the specific model that generated the pharmacokinetic parameter estimates must be appreciated. Are the parameters compatible with the model in which it will be used to make predictions? Many pharmacokinetic parameters are model-dependent, and serious errors may occur if the inappropriate parameters are used. A pharmacokinetic model is simply an artificial mathematical link to the underlying interaction of a drug’s pharmacology with an animal’s physiology (Fig. 1.1). The nature of the link will determine the types of parameters calculated.
Fig. 1.1 Conceptual framework of how a pharmacokinetic model links the observed data to the underlying biology controlling drug disposition.
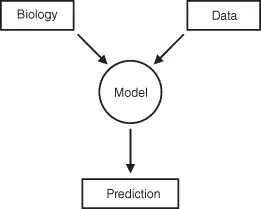
A common misconception is that if one specific model fails to adequately predict the data or experimental scenario, then the process being studied is assumed to not be amenable to pharmacokinetic analysis. Often, the fault is that insufficient data have been collected to properly define the model and its so-called inference space. The “links” were not properly constructed. In other cases, incomplete understanding of the disposition processes involved resulted in construction of a woefully inadequate model in the first place. The limitations of specific models and techniques must be appreciated before extrapolations can be made.
This book is also written for the individual who never plans on actually doing a pharmacokinetic study but desires to understand more about the time course of drug movement throughout the body. The primary goal of pharmacokinetics is to generate parameters that are mathematical abstractions that quantitate physiological processes as an aid to better understanding drug disposition. Mathematical modeling generates parameters that may vary as the physiology varies as a result of disease, age, sex, or drug-induced toxicity. The parameters are mathematical constructs that reflect changes in underlying physiology. There is no absolute value of any parameter that exists independent of the model; parameters are defined by the model and reflect the nature of the mathematical links to the physiology.
Models may be classified as mechanistic (e.g., compartmental and physiological), which represent some abstraction of the underlying physiologic reality, or as empirical (e.g., noncompartmental, neural-net analysis), which are restricted to predicting observed data. Alternatively, models may be classified as deterministic and thus purport to have exact predictability; or stochastic, which incorporate a level of statistical uncertainty in the predictions. An understanding of how models and links are derived is necessary for a thorough understanding of drug disposition and, ultimately, drug efficacy or toxicity.
There are many misconceptions as to what a pharmacokinetic study actually entails. Many workers in veterinary medicine believe that measuring drug concentrations in plasma or blood and plotting the resulting concentration–time profile comprises such a study. Similarly, some feel that if parameters such as peak concentration (Cmax), time to peak concentration, and the area under the concentration–time curve (AUC) are recorded, a pharmacokinetic analysis has been performed. In fact, to determine product bioequivalence, this does comprise a complete study. A new Chapter 15 has been added to this edition to overview the use of such approaches and present appropriate statistical techniques used in determining product bioequivalence by regulatory authorities. As will be stressed throughout this book, the problem with such analyses is that they are descriptive only for the experiment performed and are difficult to use for extrapolation to another animal or clinical conditions.
A pharmacokinetic study in the context of this book is defined as an experiment in which some type of mathematical model is fitted to the drug concentration–time profile in blood, tissue, and/or excreta. This opens the possibility of correlating model parameters to physiological processes or using them for interspecies extrapolation. In these types of analyses, parameters such as half-life (T½), volume of distribution (Vd), and clearance (Cl) are calculated in addition to the descriptive parameters mentioned above. A separate chapter will be devoted to the physiology underlying each type of parameter. Similarly, the major types of modeling paradigms adopted will be developed and compared. Whatever type of model is employed (linear vs. nonlinear, compartmental vs. noncompartmental), a model is only a tool to estimate drug concentrations and generate parameters that are useful for further analyses and quantitating the biological process under investigation. Models are neither correct nor incorrect, but should be judged only as to how accurately drug concentrations are predicted under new exposure conditions.
The selection of a model relative to its use for prediction of future events is an important decision. Fig. 1.2 depicts how three radically different mathematical models may fit the same limited data set. The three models are statistically equivalent in terms of their ability to describe the observed data, and thus all are mathematically appropriate. However, only the exponential model has a relatively direct link to biological reality. All three predict very different drug concentrations for times beyond the actual data collected. Within the observed time interval, all models accurately interpolate drug concentrations at times between collections. This is defined as the inference space of the model. However, extrapolation outside of the observed time window requires knowledge that the model has biological reality at these time points. Collection of as few as one or two additional data points would expand the model’s inference space and help select the more predictive model. It is surprising how often this simple limitation of fitting equations to data is overlooked.
Fig. 1.2 Illustration of how three very diverse mathematical models may adequately describe the same limited set of data yet result in very different values when predictions are extrapolated beyond the models’ inference space. Linear (—); sinusoidal (– –); exponential (— — —).
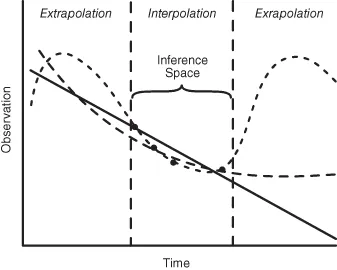
Completely independent of the model selected, it is the fitting of the model to the data, for the purposes of the investigator, that must be optimized. This is wher...
Table of contents
- Cover
- Half title page
- Title page
- Copyright page
- Coauthors
- Preface
- 1 Introduction
- 2 Principles of Drug Movement in the Body
- 3 Quantitative Structure–Permeability Relationships
- 4 Absorption
- 5 Distribution
- 6 Renal Elimination
- 7 Hepatic Biotransformation and Biliary Excretion
- 8 Compartmental Models
- 9 Noncompartmental Models
- 10 Nonlinear Models
- 11 Physiological Models
- 12 Dosage Regimens
- 13 Simultaneous Pharmacokinetic–Pharmacodynamic Modeling
- 14 Study Design and Data Analysis
- 15 Bioequivalence Studies
- 16 Population Pharmacokinetic Models
- 17 Dosage Adjustments in Disease States
- 18 Interspecies Extrapolations
- 19 Tissue Residues and Withdrawal Times
- Index
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Comparative Pharmacokinetics by Jim E. Riviere in PDF and/or ePUB format, as well as other popular books in Medicine & Veterinary Medicine. We have over 1.5 million books available in our catalogue for you to explore.