Advances in knowledge and technology have revolutionized the process of drug development, making it possible to design drugs for a given target or disease. Building on the foundation laid by the previous three editions, Smith and Williams Introduction to the Principles of Drug Design and Action, Fourth Edition includes the latest informatio

eBook - ePub
Smith and Williams' Introduction to the Principles of Drug Design and Action
- 720 pages
- English
- ePUB (mobile friendly)
- Available on iOS & Android
eBook - ePub
Smith and Williams' Introduction to the Principles of Drug Design and Action
About this book
Trusted by 375,005 students
Access to over 1.5 million titles for a fair monthly price.
Study more efficiently using our study tools.
Information
1.
PROCESSES OF DRUG HANDLING BY THE BODY
DAVID K.LUSCOMBE and PAUL J.NICHOLLS
1.1 INTRODUCTION
To be useful as a medicine, a drug must be capable of being delivered to its site of action in a concentration large enough to initiate a pharmacological response. This concentration will depend on the amount of drug administered, the rate and extent of its absorption and its distribution in the blood stream to other parts of the body. The medicine will continue to act until the concentration of drug drops below its threshold for pharmacological activity either due to its removal (excretion) from the body in an unchanged form or after its metabolism to a more polar substance. The interrelationship between the absorption, distribution, metabolism and excretion of a drug is referred to as pharmacokinetics and describes how drugs are handled by the body. Such knowledge of a new drug is fundamental to the drug development process, to enable selection of the optimal dose, route and frequency of dosing to produce the desired clinical effect, without producing unwanted side-effects.
1.2 ABSORPTION
Whilst most medicines are taken by mouth and swallowed, other routes of administration include sublingual dosing in which the drug is placed under the tongue, rectal, inhalation, application to epithelial surfaces (skin patches), and injection either intravenously, intramuscularly or subcutaneously. With the exception of the intravenous route, in which the drug is administered directly into the bloodstream, a drug must initially be absorbed from its site of administration before it can enter the bloodstream and be distributed to its various sites of action. Clearly, the process of absorption is of fundamental importance in determining the pharmacodynamic and hence the therapeutic activity of a medicine. Delays or losses of drug during absorption may contribute to variability in drug response and may even result in a drug appearing to lack clinical effectiveness in some patients. Different formulations of the same active ingredient may lead to varying rates of absorption resulting in markedly different pharmacokinetic profiles in the same patient. Since the process of absorption involves the passage of a drug across one or more cell membranes, physico-chemical characteristics such as molecular size and shape, as well as solubility of the ionized and non-ionized forms will play an essential role in determining the overall pharmacodynamic activity of a drug. A basic knowledge of the physical and chemical principles governing the active and passive transfer of drugs across biological membranes is therefore necessary.
1.2.1 Transfer of drugs across cell membranes
Living cells are surrounded by a semipermeable membrane measuring approximately 8μ in thickness. The ease with which a drug passes across such a membrane will reflect the concentration of drug achieved in the tissues and body fluids and hence at its pharmacological site of action. In general, there are four ways by which substances are able to cross cell membranes; diffusion through the lipid component of the membrane, diffusion through aqueous channels or pores in the membrane, combination with an active carrier molecule, by pinocytosis. The commonest and most important mechanism by which drugs are transferred across biological membranes is by passive diffusion. Transfer takes place along a concentration gradient from a region of higher concentration to one of low concentration following a first-order rate reaction. The greater this concentration gradient, the greater the rate of diffusion of a drug across the cell membrane. However, the ease with which a drug passes across a membrane will depend on the characteristics of both the drug molecule and the cell membrane. The drug’s partition coefficient between the lipid cell membrane and the aqueous environment is a major source of variability. Most drugs are weak acids or weak bases, existing in aqueous solution as an equilibrium mixture of non-ionized and ionized species. The non-ionized form is lipid soluble and therefore diffuses readily across cell membranes. In contrast, ionized compounds partition poorly into lipids and as a result are only slowly transported across biological membranes. In general, the higher the partition coefficient between lipid and water the more rapidly the drug is able to pass across cell membranes.
The ratio of non-ionized to ionized drug when in aqueous solution is pH-dependent and can be calculated from the general form of the Henderson-Hasselbach equation:
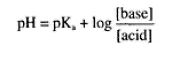
where pKa is the dissociation constant. For drugs that are weak acids, the acid form is in the non-ionized form whilst for drugs that are weak bases, the base form is non-ionized. Thus, a solution of the weak acid aspirin (pKa 3.5) in the stomach at pH 1 will have greater than 99% of the drug in the non-ionized form and consequently is lipid soluble and will be rapidly absorbed into the bloodstream. Likewise, other weak acidic drugs will be absorbed in the stomach because they exist largely in their non-ionized form at low pH values. In contrast, most basic drugs are so highly ionized in the acid content of the stomach that absorption is negligible whilst in the near neutral fluids of the small intestine the absorption of weak basic drugs such as codeine (pKa8) is rapid.
Nevertheless, it should be pointed out that the absorption of all orally administered drugs, weak acids as well as weak bases, probably takes place more rapidly in the small intestine than in the stomach. This is because the gastric mucosa has a relatively small surface area and its covering of protective mucus provides a poor site for absorption compared with the large surface area provided within the small intestine. Consequently, whilst only 0.1% of aspirin is in its non-ionized form at pH 7.0, aspirin is well absorbed from the small intestine following oral dosing. Strong organic acids and bases such as sulphonic acid derivatives and quaternary ammonium bases, are ionized over a wide range of pH values resulting in low lipid solubility and in consequence, such drugs are poorly absorbed from the gastrointestinal tract when administered orally. The pKa values for a number of acidic and basic drugs are illustrated in Figure 1.1.

Figure 1.1 pKa values of some acidic and basic drugs.
Whilst most drugs cross cell membranes by passive diffusion, some drugs such as methotrexate and 5-flourouracil are carried by an active transport mechanism which requires the expenditure of metabolic energy. The carrier is a membrane component capable of forming a complex with the drug to be transported. The complex moves across the membrane releasing the drug on the other side. Not surprisingly, carrier-aided transport systems can be saturated, thus limiting the rate of transport. This is in contrast to the process of passive diffusion across lipid membranes, or passage through pores, where the amount of drug conveyed increases proportionally with an increase in concentration. Active transport processes take place in the gastrointestinal tract (e.g. amino acids), in the renal tubules, and across membranes dividing extracellular from intracellular compartments at the blood-brain and placental barriers.
Water-soluble substances such as alcohol are able to readily diffuse through the aqueous channels or pores in cell membranes providing their molecular weights are not greater than 100–200 Da. Since most drugs fall within the molecular weight range 200–1000 Da, diffusion through these aqueous pores is unimportant for almost all substances with the exception of water, alcohol and other small polar molecules. Drug molecules can also be transported across cell membranes by an active uptake process similar to phagocytosis called pinocytosis. This involves the invagination of part of the cell membrane and the trapping of drops of extracellular fluid containing solute molecules which are thus carried through the membrane in the resulting vacuoles. Whilst this mechanism appears important in the absorption of some large molecules such as insulin which crosses the blood brain barrier by this process, pinocytosis is of little importance in the transport of small molecules across biological membranes except possibly in the case of oral vaccines. However, this process may become important if liposomes are used as a means of targeting a drug at a specific site of action since they may be taken up selectively by cells capable of pinocytosis.
1.2.2 Oral dosing
The most common route of drug administration is by swallowing. This provides a convenient, relatively safe and economical method of dosing which, subject to the drug being presented in a palatable and suitable form, is the route preferred by most patients. Normally, about 75% of a drug given orally will be absorbed in 1 to 3 hours after dosing. To be effective a drug must be stable in the acid of the stomach fluids and not cause irritation of the gastrointestinal mucosa which might induce nausea and vomiting. It should not pass too rapidly through the stomach or interact with other drugs being administered concurrently. Whether the drug is formulated as a tablet, capsule or liquid preparation the most important site for drug absorption is the small intestine because it offers a far greater epithelial surface area for drug absorption than other parts of the gastrointestinal tract. Apart from the above, many other factors influence the rate and extent of drug absorption such as the physico-chemical properties of the drug, particle size, its concentration at the absorption site and splanchnic blood flow. In fact, the intestine has an excellent blood supply which ensures that any absorbed drug is rapidly transported into the bloodstream as soon as it passes through the intestinal membrane, maintaining a concentration gradient across the membrane. For highly lipid-soluble drugs, or those that pass freely through the aqueous-filled pores, passage across a membrane may be so rapid that equilibrium is established between the drug in the bloodstream and that at the site of absorption by the time the blood is removed from the membrane. In such cases, the rate-limiting step controlling drug absorption is blood flow and not transportation across the intestinal cell membranes.
Drug absorption following oral dosing is generally favoured by an empty stomach. Food will effectively reduce the concentration of drug in the gastrointestinal tract which will limit its rate of absorption although not the total amount of drug absorbed. Furthermore, gastric emptying will be delayed slowing the onset of action of drugs such as antibiotics, analgesics and sedatives. In particular, gastric emptying is slowed by fats and fatty acids in the diet, and bulky or viscous foods. Some disorders will also slow gastric emptying, for example, mental depression, migraine, gastric ulcers and hypothyroidism whilst many drugs including propantheline, imipramine and the antacid aluminium hydroxide will all produce the same effect. In contrast, factors which promote gastric emptying will result in an increased rate of absorption of nearly all drugs. Such factors include fasting or hunger, alkaline buffer solutions, diseases such as hyperthyroidism and the anti-emetic agent, metoclopramide. Generally, the gastric emptying of liquids is much faster than that of solid food or solid dosage forms. It is for this reason, that tablets and capsules should be taken orally with at least half a glassful of water. In contrast, drugs known to irritate the gastric mucosa, for example, antiinflammatory agents, should be taken immediately after a meal, even though this may decrease its rate of absorption, as the likelihood of induced nausea will be diminished.
The term bioavailability is used to describe the proportion of orally administered drug that passes unchanged into the bloodstream. It is particularly useful because it takes into account absorption and any local metabolic degradation that takes place in the stomach and small intestine. Bioavailability is also influenced by gastrointestinal motility, gastric pH, drug solubility, the presence or absence of food in the gastrointestinal t...
Table of contents
- Cover Page
- Title Page
- Copyright Page
- Preface
- List of Contributors
- Abbreviations
- 1. Processes of Drug Handling by the Body
- 2. The Design of Drug Delivery Systems
- 3. Intermolecular Forces and Molecular Modelling
- 4. Drug Chirality and Its Pharmacological Consequences
- 5. Quantitative Structure-Activity Relationships and Drug Design
- 6. From Programme Sanction to Clinical Trials: A Partial View of the Quest for Arimidex™, A Potent, Selective Inhibitor of Aromatase
- 7. Pro-Drugs
- 8. Design of Enzyme Inhibitors as Drugs
- 9. The Chemotherapy of Cancer
- 10. Neurotransmitters, Agonists and Antagonists
- 11. Design of Antimicrobial Chemotherapeutic Agents
- 12. Recombinant DNA Technology: Monoclonal Antibodies
- 13. Bio-Inorganic Chemistry and Its PharmaceuticaL Applications
Frequently asked questions
Yes, you can cancel anytime from the Subscription tab in your account settings on the Perlego website. Your subscription will stay active until the end of your current billing period. Learn how to cancel your subscription
No, books cannot be downloaded as external files, such as PDFs, for use outside of Perlego. However, you can download books within the Perlego app for offline reading on mobile or tablet. Learn how to download books offline
Perlego offers two plans: Essential and Complete
- Essential is ideal for learners and professionals who enjoy exploring a wide range of subjects. Access the Essential Library with 800,000+ trusted titles and best-sellers across business, personal growth, and the humanities. Includes unlimited reading time and Standard Read Aloud voice.
- Complete: Perfect for advanced learners and researchers needing full, unrestricted access. Unlock 1.5M+ books across hundreds of subjects, including academic and specialized titles. The Complete Plan also includes advanced features like Premium Read Aloud and Research Assistant.
We are an online textbook subscription service, where you can get access to an entire online library for less than the price of a single book per month. With over 1.5 million books across 990+ topics, we’ve got you covered! Learn about our mission
Look out for the read-aloud symbol on your next book to see if you can listen to it. The read-aloud tool reads text aloud for you, highlighting the text as it is being read. You can pause it, speed it up and slow it down. Learn more about Read Aloud
Yes! You can use the Perlego app on both iOS and Android devices to read anytime, anywhere — even offline. Perfect for commutes or when you’re on the go.
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Please note we cannot support devices running on iOS 13 and Android 7 or earlier. Learn more about using the app
Yes, you can access Smith and Williams' Introduction to the Principles of Drug Design and Action by H. John Smith,Hywel Williams in PDF and/or ePUB format, as well as other popular books in Medicine & Biochemistry in Medicine. We have over 1.5 million books available in our catalogue for you to explore.